Генетические причины замершей беременности, или зачем знать свой кариотип
Содержание:
- Признаки
- Нарушения в наборах хромосом
- Аутосомные болезни
- Прогнозы
- Сущность заболеваний
- Связанные нарушения
- Хромосомные аномалии плода – какие могут быть исходы
- Патофизиология
- Расшифровка маркеров крови
- Аномалии половых хромосом
- Генетические анализы при подозрении наличия хромосомных аномалий
- Расшифровка анализа
- Хромосомные болезни
- 2.Синдром Патау
- Причины хромосомных аномалий у детей
- Нарушения структуры хромосом[править | править код]
- Двойной и тройной тесты
Признаки
Процесс развития аномалий во внутриутробном состоянии сегодня изучен недостаточно. Именно поэтому признаки аномалий считаются условными. Среди них:
- на раннем сроке беременности тянущая боль внизу живота;
- угроза выкидыша;
- нестандартная длина носовых костей;
- низкий уровень АФП и РАРР-А, а также повышенный уровень ХГЧ. Чтобы посмотреть эти показатели, в 12 недель беременной женщине назначают анализ – кровь из вены;
- неактивность плода;
- замедленное развитие трубчатых костей;
- шейная складка большего размера, чем норма;
- лоханки почек имеют увеличенные размеры;
- гипоксия;
- многоводие;
- маловодие;
- допплерометрия и КТГ с плохими показателями;
- большой мочевой пузырь;
- гидронефроз;
- наличие кист в головном мозге;
- гиперэхогенный кишечник;
- деформации лица;
- кисты в области пуповины;
- отёчность шеи и спины.
Все эти признаки могут быть и нормой развития плода, при условии подобной особенности организма ребёнка или же матери. Максимально точно убедиться в том, что присутствуют хромосомные аномалии, помогут анализы кров, инвазивные методики и УЗИ.
Нарушения в наборах хромосом
Иногда количество пар не соответствует стандарту. Проблему во внутриутробном развитии может заметить только генетик, если будущая мама добровольно пройдет исследование. Если количество нарушено, то выделяют такие заболевания:
- Синдром Клайнфельтера.
- Болезнь Дауна.
- Синдром Шерешевского-Тернера.
Консервативных методов для восполнения недостающего генетического ряда не существует на сегодняшний день. То есть подобный диагноз считается неизлечимым. Если проблема была диагностирована во время беременности, лучше всего ее прервать. В противном случае появляется больной ребенок с возможными внешними уродствами.
Болезнь Дауна
Через некоторое время болезнь
При синдроме Дауна к 21 паре прикрепляется еще одна. То есть, общее количество составляет не 46, а 47 хромосом. Патология формируется спонтанно, а ее причиной может быть сахарный диабет, пожилой возраст родителей, повышенная доза радиации, наличие некоторых хронических заболеваний.
Внешне такой ребенок отличается от здоровых сверстников. У него узкий и широкий лоб, объемный язык, большие уши, сразу бросается в глаза умственная отсталость. Также у пациента диагностируются другие проблемы со здоровьем, которые затрагивают многие внутренние системы и органы.
По большому счету хромосомный ряд будущего малыша сильно зависит от генома его матери. Именно поэтому перед началом планирования беременности необходимо пройти полноценное клиническое обследование. Оно позволит определить скрытые проблемы. Если врачи не обнаружат противопоказаний, можно думать о зачатии ребенка.
Синдром Патау
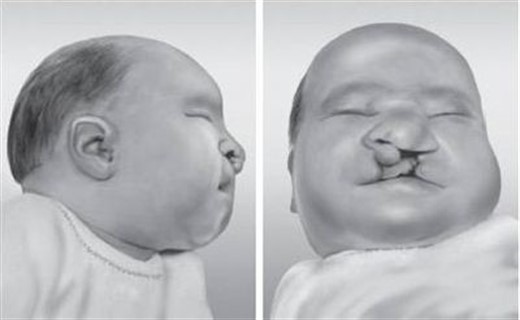
При этом нарушении наблюдается трисомия в тринадцатой паре структурных единиц. Такое заболевание встречается намного реже, чем синдром Дауна. Оно возникает, если присоединяется лишняя структурная единица или нарушается структура хромосом и их перераспределение.
Существует три основных симптома, по которым диагностируют данную патологию:
- Уменьшенные размеры глаз или микрофтальм.
- Увеличенное количество пальцев (полидактилия).
- Расщелина неба и губы.
При таком заболевании около 70% младенцев вскоре после рождения (до трех лет) умирают. Часто у детей с синдромом Патау диагностируют пороки сердца, а также головного мозга, проблемы со многими внутренними органами.
Синдром Эдвардса
Эта патология характеризуется наличием трех хромосом
Чтобы не допустить развития патологии, рекомендовано всем родителям, которые решают зачать ребенка после 35 лет, пройти специальные обследования. Также большая вероятность развития заболеваний у тех, чьи родители имели проблемы со щитовидной железой. Гуманитарии кто это читайте у нас на сайте
Аутосомные болезни
Синдром
Дауна – 47, XY + 21 или
47, XX + 21
(трисомия
по 21-й паре хромосом).
Частота – 15 случаев
на 10000 новорожденных обоих полов.
Укороченные
конечности, маленькая головка, плоское
лицо, широкая переносица, монголоидный
разрез глаз, большой язык, не помещающийся
во рту. Аномалии строения внутренних
органов. Резко выраженная умственная
отсталость. Женщины иногда могут иметь
детей, мужчины – никогда. 31% больных
умирает до 1 года, причем от обычных
простых заболеваний, так как снижен
иммунитет. Вообще живут недолго.
Иногда
синдром Дауна обусловлен не трисомией,
а транслокацией
(реципрокной) между 13 – 15 и 21 хромосомами;
в этом случае хромосом 46.
Синдром
Эдвардса – 47, ХХ + 18 или
47, XY
+ 18 (трисомия
по 18-й паре хромосом).
Частота – 1 случай
на 7000 новорожденных обоего пола.
Фенотип: множественные
аномалии – деформация черепа, «птичий
профиль» лица, короткие глазные щели,
микрофтальм, низко расположенные и
деформированные ушные раковины, короткая
шея и грудина, врожденный вывих бедра,
синдактилия, врожденные пороки сердца
и крупных сосудов. Живут недолго: 30%
умирает на первом и 50% – на втором месяце
жизни
Синдром
Патау – 47, XY
+ 13 или
47, ХХ +
13 (трисомия
по 13-й паре хромосом).
Частота
– 1 случай на 6000 новорожденных обоего
пола.
Множественные
уродства (микрофтальм, расщелина губы
и неба, череп неправильной конфигурации,
скошенный узкий лоб, плоский и широкий
нос, запавшее переносье, низко расположенные
уши, полидактилия, пороки развития
внутренних органов). Живут несколько
дней или недель.
Синдром
Лежьена – 46, ХХ, 5р- или
46, XY,
5р- (делеция
короткого плеча ( р ) 5-й хромосомы).
Частота
– 1 случай на 50000 новорожденных обоего
пола. Его еще называют «синдром
кошачьего крика».
Характеризуются
многочисленными пороками развития
черепа, строения гортани (в связи с чем
при рождении вместо плача издают звук,
похожий на кошачье мяуканье), конечностей,
сердца, почек, глаз, тяжелой формой
слабоумия. Живут такие больные недолго.
Прогнозы
Родителям, у ребёнка которых внутриутробно были обнаружены хромосомные патологии, должны понять и принять как данность, что они не лечатся. Всё, что может предложить им медицина в таком случае, — это искусственное прерывание беременности. Прежде чем принимать такое ответственное решение, нужно проконсультироваться у врачей по следующим вопросам:
- Какая именно патология была диагностирована?
- Какие последствия она будет иметь для жизни и здоровья ребёнка?
- Велика ли угроза выкидыша и мертворождения?
- До скольки лет доживают дети с таким диагнозом?
- Готовы ли вы стать родителями ребёнка-инвалида?
Чтобы принять правильное решение о том, оставить больного малыша или нет, нужно объективно оценить все возможные последствия и результаты хромосомной патологии плода совместно с врачом. Во многом они зависят от того, какую именно генетическую аномалию предполагают медики. Ведь их достаточно много.
Сущность заболеваний

Если у родителей (у обоих) имеются в роду наследственные заболевания, им в первую очередь необходимо знать, что это такое — хромосомные патологии плода, которые могут выявить у их ребёнка, пока он ещё в утробе. Осведомлённость позволит избежать нежелательного зачатия, а если это уже произошло, — исключить самые тяжёлые последствия, начиная от внутриутробной гибели малыша и заканчивая внешними мутациями и уродствами после его рождения.
У нормального, здорового человека хромосомы выстраиваются в 23 пары, и каждая отвечает за какой-то определённый ген. Всего получается 46. Если их количество или строение иное, говорят о хромосомных патологиях, разновидностей которых в генетике очень много. И каждая из них влечёт за собой опасные последствия для жизни и здоровья малыша. Основные причины такого рода аномалий неизвестны, однако существуют определённые группы риска.
Связанные нарушения
Симптомы следующих расстройств могут быть сходными с симптомами синдрома Тернера. Сравнения полезны для дифференциальной диагностики.
Синдром Нунана – генетическое заболевание, которое проявляется при рождении (врожденный). Расстройство характеризуется широким спектром симптомов и физических особенностей, которые сильно различаются по диапазону и степени тяжести.
У многих затронутых лиц ассоциированные аномалии включают:
- характерный внешний вид лица;
- широкую или перепончатую шею;
- низкую линию волос;
- типичную деформацию грудной клетки;
- низкий рост.
Характерные аномалии головной и лицевой (черепно-лицевой) области включают:
- широко расставленные глаза (глазной гипертелоризм);
- складки кожи, покрывающие внутренние углы глаз (эпикантальные складки);
- опущенные верхние веки (птоз);
- маленькую челюсть (микрогнатия);
- продавленный корень носа;
- короткий нос с широким основанием;
- низкорасположенные уши (pinnae).
Присутствуют отличительные скелетные мальформации, такие как аномалии грудины, искривление позвоночника (кифоз, сколиоз), наружное отклонение локтей (cubitus valgus). У многих младенцев с синдромом Ноонана есть дефекты сердца, такие как обструкция кровотока из нижней правой камеры в легкие (легочный клапанный стеноз).
Дополнительные аномалии включают пороки развития сосудов, лимфы, плохое свертывание крови, дефицит тромбоцитов, трудности обучения, умеренную интеллектуальную инвалидность, крипторхизм на первом году жизни у пораженных мужчин и другие симптомы.
Узнать больше Симптомы и лечение синдрома вискотта олдрича у детей
Синдром Нунан является аутосомно-доминантным генетическим расстройством, вызванным аномалиями (мутациями) четырех основных генов. Некоторые симптомы могут внешне напоминать симптомы синдрома Тернера (низкий рост, перепончатая шея). Однако между этими двумя расстройствами существует много важных различий.
Синдром нунана поражает как мужчин, так и женщин, существует нормальный хромосомный кариотип. Только женщины подвержены влиянию синдрома Тернера, который характеризуется нарушениями, влияющими на Х-хромосому.
Хромосомные аномалии плода – какие могут быть исходы
В большинстве случаев природа сама производит естественный отбор, прекращая развитие нежизнеспособного эмбриона. Хромосомные нарушения обуславливают 50-60% репродуктивных потерь (самопроизвольные аборты в сроке до 8-10 недель и не менее 10% всех случаев внутриутробной гибели ребенка). Однако вовремя не выявленные врожденные дефекты являются одной из причин рождения больного ребенка (0.4% от всех родов). В среднем на 10 миллионов населения ежегодно рождается около 3 тысяч детей с различными вариантами врожденной и наследственной патологией. Длительность жизни инвалидов с детства – не более 35 лет. Ни одна будущая мама не хочет, чтобы родился больной малыш
Ни одной семейной паре не пожелаешь такой участи, поэтому важно загодя позаботится о будущем, соблюдая рекомендации врача по прегравидарной подготовке и неинвазивному пренатальному скринингу
Патофизиология
Кариотип (хромосомный набор) человека с синдромом Дауна. В 21-й паре три хромосомы вместо двух
Синдром Дауна — хромосомная патология, характеризующаяся наличием дополнительных копий генетического материала 21-й хромосомы, либо целой хромосомы (трисомия), либо её участков (например, за счёт транслокации). Последствия от наличия дополнительной копии сильно различаются в зависимости от количества дополнительного генетического материала, генетического окружения и чистой случайности. Синдром Дауна встречается как у людей, так и у других видов (например, был обнаружен у обезьян и мышей). В 2005 году британские исследователи получили анеуплоидных трансгенных мышей с наличием 21-й человеческой хромосомы в дополнение к стандартному набору мышей. Нормальный человеческий кариотип содержит 46 хромосом и обозначается 46,XY у мужчин и 46,XX у женщин, в то время как у носителей синдрома Дауна с трисомией по 21-й хромосоме кариотип содержит 47 хромосом.
Трисомия
Трисомия — это наличие трёх гомологичных хромосом вместо пары в норме.
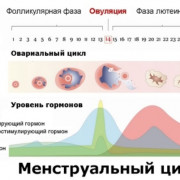
Риск рождения ребёнка с синдромом Дауна и другими численными хромосомными аномалиями растёт с возрастом матери. Точная причина этого неизвестна, но, по-видимому, она связана с возрастом яйцеклеток матери.
Трисомия происходит из-за нерасхождения хромосом во время мейоза, в результате чего возникает гамета с 24 хромосомами. При слиянии с нормальной гаметой противоположного пола образуется зигота с 47 хромосомами, а не 46-ю, как без трисомии.
Трисомия 21-й хромосомы в 95 % случаев является причиной возникновения синдрома Дауна, и в 88 % случаев из-за нерасхождения материнских гамет и в 8 % — мужских.
Мозаицизм
Трисомия обычно вызвана нерасхождением хромосом при формировании половых клеток родителя (гамет), в этом случае все клетки организма ребёнка будут нести аномалию. При мозаицизме же нерасхождение возникает в клетке зародыша на ранних стадиях его развития, в результате чего нарушение кариотипа затрагивает только некоторые ткани и органы. Данный вариант развития синдрома Дауна называется «мозаичный синдром Дауна» (46, XX/47, XX, 21). Данная форма синдрома является, как правило, более лёгкой (в зависимости от обширности изменённых тканей и их расположения в организме), однако более трудна для пренатальной диагностики.
По данному типу синдром появляется в 1—2 % случаев.
Робертсоновские транслокации
Дополнительный материал 21-й хромосомы, вызывающий синдром Дауна, может появиться вследствие наличия робертсоновской транслокации в кариотипе одного из родителей. В данном случае длинное плечо 21-й хромосомы прикреплено к плечу другой хромосомы (чаще всего 14-й ). Фенотип у человека с робертсоновскими транслокациями соответствует норме. Во время репродукции нормальный мейоз повышает шанс на трисомию 21-й хромосомы и рождения ребёнка с синдромом Дауна. Транслокации с синдромом Дауна часто называют семейный синдром Дауна. Эта форма не зависит от возраста матери. Данный тип появления синдрома занимает 2—3 % от всех случаев.
Дупликация части хромосомы 21
Очень редко участки 21-й хромосомы могут быть удвоены в результате хромосомной перестройки. При этом возникают дополнительные копии некоторых, но не всех генов из 21-й хромосомы. Если продублируются фрагменты, обусловливающие физические и психологические проявления синдрома Дауна, то ребёнок родится с этим синдромом. Такие хромосомные перестройки происходят крайне редко, и не существует оценки периодичности данного явления.
Формы синдрома Дауна
Примерно в 91 % случаев возникает ненаследственный вариант синдрома — простая полная трисомия 21 хромосомы, обусловленная нерасхождением хромосом во время мейоза. Примерно у 5 % людей с синдромом Дауна наблюдается мозаицизм (не все клетки содержат лишнюю хромосому). В остальных случаях синдром вызван спорадической или наследуемой транслокацией 21-й хромосомы. Как правило, такие транслокации возникают в результате слияния центромеры 21-й хромосомы и другой акроцентрической хромосомы. Фенотип больных определяется трисомией 21q22. Повторный риск рождения ребёнка с синдромом Дауна у родителей с нормальным кариотипом составляет около 1 % при обычной трисомии у ребёнка.
Информация об этих редких формах значима для родителей, так как риск рождения других детей с синдромом Дауна различен при разных формах. Тем не менее, для понимания развития детей эти различия не так важны. Хотя профессионалы склонны считать, что дети с мозаичной формой синдрома Дауна отстают в своём развитии меньше детей с другими формами этого синдрома, достаточно убедительных сравнительных исследований на эту тему пока нет.
Расшифровка маркеров крови
Уровень ХГЧ
Хорионический гонадотропин человека включает две субъединицы – альфа и бета. Уникальный свободный бета-ХГЧ является биохимическим маркером.
| Неделя беременности | Норма свободного бета-ХГЧ (нг/моль) |
| 10 | 25,8-181,6 |
| 11 | 17,4-130,4 |
| 12 | 13,4-128,5 |
| 13 | 14,2-114,7 |
| 14 | 8,9-79,4 |
Повышение уровня свободного бета-ХГЧ может свидетельствовать о таких явлениях:
- синдром Дауна (превышение нормы в два раза);
- многоплодие;
- сахарный диабет у беременной;
- гестоз (повышение давления, отечности, белок в моче);
- аномальное развитие плода;
- хориокарцинома (злокачественная опухоль, которая образуется из клеток плода);
- пузырный занос (развитие плода нарушается, ворсины хориона разрастаются в пузыри).
Низкий уровень свободного бета-ХГЧ иногда говорит о:
- синдроме Эдвардса, синдроме Патау;
- задержке развития;
- угрозе выкидыша;
- хронической плацентарной недостаточности.
Уровень PAPP
РАРР-А – протеин-А плазмы. Отклонения от нормы зачастую указывают на пороки развития. Считается, что после 14 недели анализ на РАРР-А более не информативен.
| Неделя беременности | РАРР (мЕд/мл) |
| 10-11 | 0,32-2,42 |
| 11-12 | 0,46-3,73 |
| 12-13 | 0,7-4,76 |
| 13-14 | 1,03-6,01 |
Понижение уровня РАРР-А может указывать на:
- многоплодие;
- низкое расположение плаценты;
- большие размеры плода или плаценты.
Понижение уровня РАРР-А характерно при:
- синдроме Дауна, синдроме Эдвардса, синдрома Патау, синдроме Корнелии де Ланге;
- выкидыше, гибели плода;
- преэклампции (тяжелая степень гестоза, когда артериальное давление повышается до критических отметок);
- фетоплацентарной недостаточности, гипотрофии плода (из-за нехватки питания снижается масса тела ребенка).
Обычно эти показатели изучают совместно. При снижении уровня РАРР-А и повышении ХГЧ есть риск возникновения синдрома Дауна, а при нехватке обоих – синдрома Патау или синдрома Эдвардса.
Уровень АФП
Альфа-фетопротеин – белок, который выделяется желточным мешком плода в начале беременности и печенью под конец. АФП также синтезируется в желтом теле яичников женщины до 5-й недели. Уровень белка разнится для отдельных периодов беременности.
Роль АФП заключается в транспортировке белков и жиров от матери ребенку, поддержании давления в сосудах плода, мешает гормонам матери повлиять на него. Также АФП играет важную роль в осуществлении иммуносупресии между матерью и ребенком (подавление выработки антител иммунитетом матери на неизвестный организм).
| Неделя беременности | Концентрация АФП (МЕ/мл) |
| 1-13 | 0,5-15 |
| 14-16 | 15-60 |
| 17-20 | 15-95 |
| 21-24 | 27-125 |
| 25-28 | 52-140 |
| 29-30 | 67-150 |
| 31-32 | 100-250 |
| 33-42 | показатель не информативен |
Уровень эстрадиола
Во втором триместре посредством анализа крови выявляют также уровни ингибина А, плацентарного лактогена и неконъюгированного эстрадиола. Подсчет результатов совершается компьютером.
| Результат | Вероятность хромосомных патологий |
| 1:100 | очень высокая |
| 1:1000 | норма, при заниженном показателе могут быть аномалии развития |
| 1:10000 | низкая |
При результате ниже 1:400 тест проводят второй раз. Если показатели выше, женщина может спокойно доносить малыша.
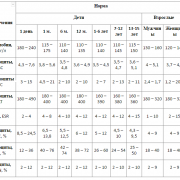
Расшифровка маркера по росту трубчатых костей
| Неделя беременности | Бедренная кость | Кость голени | Плечевая кость | Кости предплечья (локтевая и лучевая) |
| 11-12 | 3,4-4 | |||
| 13-14 | 7-9 | |||
| 15-16 | 13-17 | 15 | 15 | 12 |
| 17-18 | 20-23 | 17-20 | 17-20 | 15-17 |
| 19-20 | 26-29 | 23-26 | 23-26 | 20-22 |
| 21-22 | 32-26 | 29-31 | 29-31 | 24-26 |
| 23-24 | 37-40 | 34-36 | 34-36 | 29-31 |
| 25-26 | 42-45 | 37-41 | 39-41 | 33-35 |
| 27-28 | 47-49 | 43-45 | 43-45 | 37-39 |
| 29-30 | 50-52 | 47-49 | 47-49 | 40-42 |
| 31-32 | 54-56 | 50-51 | 51-52 | 44-45 |
| 33-34 | 58-60 | 53-33 | 54-55 | 46-48 |
| 35-36 | 62-64 | 56-57 | 57-58 | 49-50 |
| 37-38 | 66-68 | 59-60 | 59-60 | 51-52 |
| 39-40 | 69-70 | 61-62 | 60-61 | 53-54 |
Аномалии половых хромосом
Наиболее частыми аномалиями половых хромосом является синдром Тернера (моносомия Х, или 45, Х0, мозаицизм) и синдром Кляйнфельтера (47, ХХУ). Это может быть обусловлено тем обстоятельством, что кариотипы 47, ХХХ и 47, ХVV не имеют выраженных фенотипических различий и поэтому идентифицируются реже.
Синдром Тернера (Шерешевского — Тернера) может быть вызван потерей родительской хромосомы, мозаицизмом (45, Х / 46, ХХ или 45, Х / 46, ХУ) или структурными аномалиями Х-хромосомы (делеции, изохромосомы).
Индивиды с синдромом Тернера имеют женский фенотип, дисгенезию гонад, низкий рост, первичные, отсутствие вторичных половых признаков, крыловидные складки шеи, низко расположенные уши, низкую заднюю границу роста волос, дискообразную грудную клетку с широкой расстоянием между сосками, аномалии почек, лимфедему конечностей при рождении и кардиоваскулярные аномалии, чаще коарктацию аорты. Но при ультразвуковом исследовании может проявляться только одна аномалия — кистозная гигрома. Скрининг-теста синдромом Тернера еще не существует, и поэтому частота рецидивов не определена.
В случае синдрома Кляйнфельтера развитие яичек сначала является нормальным. Но присутствие не менее 1 лишней Х-хромосомы приводит к гибели зародышевых клеток на этапе их поступления в мейоз, что приводит к уменьшению яичек и гиалинизации семенных протоков. Итак, классические симптомы синдрома Кляйнфельтера включают бесплодие, гинекомастию, задержку умственного развития, повышения уровня гонадотропинов вследствие уменьшения уровня циркулирующих андрогенов. Скрининг-теста по выявлению синдрома Кляйнфельтера также не существует, следовательно, пренатальный диагноз этих хромосомных аномалий возможно только при использовании биопсии хориона или амниоцентеза.
Генетические анализы при подозрении наличия хромосомных аномалий
На основании заключения УЗИ или при неблагоприятных результатах биохимического скрининга генетик может предложить будущей маме пройти инвазивное исследование. В зависимости от срока это может быть биопсия хориона или плаценты, амниоцентез или кордоцентез. Такое исследование дает высокоточные результаты, но в 0,5% случаев такое вмешательство может стать причиной выкидыша.
Забор материала для генетического исследования проводят под местной анестезией и при УЗИ-контроле
Тонкой иглой врач делает прокол матки и осторожно берет генетический материал. В зависимости от срока беременности это могут быть частицы ворсин хориона или плаценты (биопсия хориона или плаценты), амниотическая жидкость (амниоцентез) или кровь из пуповиной вены (кордоцентез)
Полученный генетический материал оправляют на анализ, который позволит определить или исключить наличие многих хромосомных аномалий: синдром Дауна, синдром Патау, синдром Эвардса, синдром Тернера (точность – 99%) и синдром Клайнфельтера (точность — 98%).
Делать этот тест можно начиная с 10 недели беременности
Важно понимать, что этот тест пока мало распространен в России, его делают очень немногие клиники, и далеко не все врачи считаются с его результатами. Поэтому нужно быть готовыми к тому, что врач может настоятельно рекомендовать инвазивное обследование в случае высоких рисков по УЗИ или биохимическому скринингу
Как бы там ни было – решение всегда остается за будущими родителями.
В нашем городе неинвазивные пренатальные генетические тесты делают клиники:
-
«Авиценна». Тест Panorama. Неинвазивная пренатальная генетическая диагностика анеуплоидий 42 т.р. Неинвазивная пренатальная генетическая диагностика анеуплоидий и микроделеций — 52 т.р
-
«Алмита». Тест Panorama. Стоимость от 40 до 54 т.р. в зависимости от полноты исследования.
-
«УЗИ-студия». Тест Prenetix. Стоимость 38 т.р.
Расшифровка анализа
Результаты анализа расшифровывает, в первую очередь, акушер-гинеколог, ведущий беременность; получение нормальных или низких показателей риска не требует специальных знаний для расшифорвки. Если же результаты тестов оказались неблагоприятными, генетик и врач-акушер-гинеколог обязаны тщательно изучить результаты пренатального скрининга и согласовать их с семейной парой; специалисты информируют родителей о возможных рисках. При необходимости генетик назначает консультацию врача, который занимается лечением конкретной генетической аномалии, или врача-перинатолога, который ведет сложные беременности. Помощь специалиста позволяет будущим родителям подготовиться к рождению ребенка и изучить все тонкости постнатального периода.
Методами инвазивного (нехирургического) обследования беременных являются: биопсия хориона (предшественника плаценты), амниоцентез, кордоцентез (взятие крови из пуповины), а также редкие — биопсия кожи плода и эмбриоскопия. Их используют только в случае высокой вероятности развития генетического дефекта и аномалий.
Ультразвуковые маркеры
Врач ультразвуковой диагностики должен быть предупрежден о полученных результатах биохимического скрининга (и предыдущих ультразвуковых исследований)
Специалист УЗИ обращает внимание на размер воротниковой складки, копчиково-теменной размер, частоту сердцебиения плода, состояние лицевого скелета, длины трубчатых костей, состояние мочевого пузыря, почек, кишечника, количество околоплодных вод. Любые отклонения в ультразвуковой картине могут говорить о наличии генетической патологии и должны быть оценены ведущим беременность акушером-гинекологом и генетиком.
Маркеры крови
При стандартном скрининге берут два показателя крови:
-
Ассоциированный с беременностью протеин-А (PAPP-A);
-
Свободный хорионический гонадотропин (бетта-фракция).
Эти показатели зависят от срока беременности; диапазон нормы широкий и зависит от многих обстоятельств, в т.ч. от региона проживания. При расчете учитываются данные анамнеза, и такой расчет позволяет более точно определить вероятность патологии плода.
Хромосомные болезни
Об их проявлении говорят при обнаружении тяжёлых врождённых генетически обусловленных заболеваний, проявляющихся врождёнными пороками развития. Такие болезни свидетельствуют о наиболее масштабных изменениях, произошедших в ДНК.
Сбой может возникнуть на любом этапе, даже в момент зачатия, при слиянии нормальных родительских клеток. Учёным пока ещё не удаётся влиять на этот механизм и предотвращать его. Вопрос этот изучен не до конца.
Для человека хромосомные мутации чаще носят негативный характер, что проявляется в возникновении выкидышей, мертворождении, проявлении уродств и отклонений в интеллекте, появлении генетически обусловленных опухолей. Все подобные болезни условно делят на 2 группы:
- Те, что вызваны нарушением числа хромосом в геноме. Эти аномалии составляют львиную долю всех хромосомных болезней. Причины, вызывающие их, – моносомии, трисомии и другие количественные нарушения. В эту же группу входят триплоидии и тетраплоидии, вызывающие гибель плода в утробе и его нежизнеспособность, приводящую к младенческой смертности. Самой распространённой и хорошо изученной болезнью этой группы является синдром Дауна, возникающий из-за трисомии или транслокации 21-й хромосомы.
- Те, что обусловлены изменениями в структуре самих хромосом. В этом случае происходит частичная дупликация или делеция хромосомных участков. Это влечёт за собой умственную отсталость, задержку роста и развития, характерные внешние проявления, врождённые пороки внутренних органов. Изменения могут коснуться и числа половых хромосом. У таких больных впоследствии выявляется бесплодие.
2.Синдром Патау
Частота
встречаемости 1: 6000 новорождённых.
Различают три формы:
—
простая трисомия по хромосоме 13 (75 %
случаев);
—
транслокация (чаще робертсоновская)
(20 % случаев);
—
мозаичная (5%).
Фенотипы:
микроцефалия, тригоноцефалия (расширение
черепа в затылочной и сужение в лобной
части), узкие глазные щели, широкое
основание носа, низко посаженные
деформированные уши, микрофтальмия
(малые размеры глазного яблока),
микрогнатия (малые размеры верхней
челюсти), расщелина губы и нёба,
полидактилия, пороки внутренних органов
(головного мозга, сердца и сосудов,
почек, пищеварения, половых органов).
Дети погибают обычно в течение первых
трёх месяцев жизни.
Причины хромосомных аномалий у детей
Среди причин возникновения аномалии следует назвать:
- генетические факторы; кроме того — какие-либо нарушения эмбрионального развития (вследствие воздействия на развивающийся эмбрион механических, химических, температурных факторов, рентгеновских лучей и других повреждающих моментов;
- вследствие плохого питания эмбриона);
- довольно часто причиной развития аномалии становится внутриутробная инфекция.
Хромосомные аномалии плода
Примерами аномалий, развившихся вследствие внутриутробной инфекции, могут служить микроцефалия, гидроцефалия, врожденная слепота. Те аномалии, что выражены резко, называют пороками развития или уродствами.
Аномалии в развитии детей могут быть не только морфологические, но и функциональные — так называемые аномалии конституции; при таких аномалиях физиологическое состояние организма определяют как нестойкое.
Нарушения структуры хромосом[править | править код]
- Транслокации — обменные перестройки между негомологичными хромосомами.
- Делеции — потери участка хромосомы. Например, синдром кошачьего крика связан с делецией короткого плеча 5-й хромосомы. Признаком его служит необычный плач детей, напоминающий мяуканье или крик кошки. Это связано с патологией гортани или голосовых связок. Наиболее типичным, помимо «кошачьего крика», является умственное и физическое недоразвитие, микроцефалия (аномально уменьшенная голова).
- Инверсии — повороты участка хромосомы на 180 градусов.
- Дупликации — удвоения участка хромосомы.
- Изохромосомия — хромосомы с повторяющимся генетическим материалом в обоих плечах.
- Возникновение кольцевых хромосом — соединение двух концевых делеций в обоих плечах хромосомы.
В настоящее время[когда?] у человека известно более 700 заболеваний, вызванных изменением числа или структуры хромосом. Около 25 % приходится на аутосомные трисомии, 46 % — на патологию половых хромосом. Структурные перестройки составляют 10,4 %. Среди хромосомных перестроек наиболее часто встречаются транслокации и делеции.
Двойной и тройной тесты
Большая часть беременных в Российской Федерации, согласно стандартам, сдает первый генетический тест (так называемый первый скрининг) для исключения серьёзной хромосомной патологии плода. В число диагностируемых заболеваний входят синдромы Дауна, Эдвардса и Патау; тест проводят на сроке 10-12 недель беременности, чтобы дать возможность прервать беременность. Это так называемый двойной тест – пациентка сдаёт биохимический анализ крови и ей проводят ультразвуковое исследование (измеряется т.н. «воротниковая зона»). На основании полученных результатов рассчитывается вероятность рождения ребенка с хромосомной патологией.
Расчет может быть проведен с учетом нескольких (трех и даже четырех) показателей, в 16-18 недель. Обязательная сдача таких тестов отменена из-за низкой диагностической ценности, однако ультразвуковой скрининг в этом сроке мы в нашей клинике проводим. Отдельно следует упомянуть расширенные тесты, которые можно проводить уже с 9 недель беременности. Такие тесты могут давать информацию по большинству хромосом плода, сравниваясь по ценности и точности диагностики с инвазивными тестами. Существуют тесты, которые могут работать при суррогатном материнстве, при донорских программах и даже при двойнях (стандартные скрининги не дают таких возможностей).