Вылечить миодистрофию дюшенна: конкуренция групп, единство методик
Содержание:
- Симптомы Прогрессирующей мышечной дистрофии Дюшенна:
- Разновидности мышечной дистрофии
- Классификация миопатий
- Диагностика
- Симптомы миопатий
- Причины генетического расстройства
- Что такое миопатия Дюшена
- Что это такое
- Диагностические мероприятия
- Киста Беккера — что это такое?
- Управление на ранней стадии — когда ребенок гуляет, до 11 лет
- Лечение миодистрофии Эрба-Рота
- Признаки и симптомы мышечной дистрофии Дюшенна
- Классификация миопатий
- Дальнейшее чтение и ссылки
- Профилактика и прогноз
Симптомы Прогрессирующей мышечной дистрофии Дюшенна:
Признаки заболевания проявляются в первые 1–3 года жизни
Уже на 1-м году обращает на себя внимание отставание детей в моторном развитии. Они, как правило, с задержкой начинают садиться, вставать, ходить
Движения неловкие, при ходьбе дети не усто йчивы, часто спотыкаются, падают. В 2–3 года по являются мышечная слабость, патологическая мышечная утомляемость, проявляющаяся при физической нагрузке – длительной ходьбе, подъеме на лестницу, изменение походки по типу «утиной». В этот период обращает на себя внимание своеобразная «стереотипная» динамика движений детей во время вставания из горизонтального положения, из положения на корточках или со стула. Вставание происходит поэтапно, с активным использованием рук – «взбирание лесенкой» или «взбирание по самому себе». Атрофии мышц всегда симметричны. Вначале они локализуются в проксимальных группах мышц нижних конечностей – мышцах тазового пояса, бедер, а через 1– 3 года быстро распространяются в восходящем направлении на проксимальные группы мышц верхних конечностей – плечевой пояс, мышцы спины. Вследствие атрофии появляются лордоз, «крыловидные» лопатки, «осиная» талия. Типичным, «классическим» симптомом заболевания является псевдогипертрофия икроножных мышц.
При пальпации мышцы плотны, безболезненны. У многих больных в результате селективного и неравномерного поражения различных групп мышц рано возникают мышечные контрактуры и сухожильные ретракции. Мышечный тонус снижен преимущественно в проксимальных группах мышц. Сухожильные рефлексы изменяются с различной последовательностью. В ранних стадиях болезни исчезают коленные рефлексы, позднее – рефлексы с двуглавой и трехглавой мышц. Пяточные (ахилловы) рефлексы длительное время остаются сохранными.
Снижение амплитуды осцилляции и увеличение полифазности.
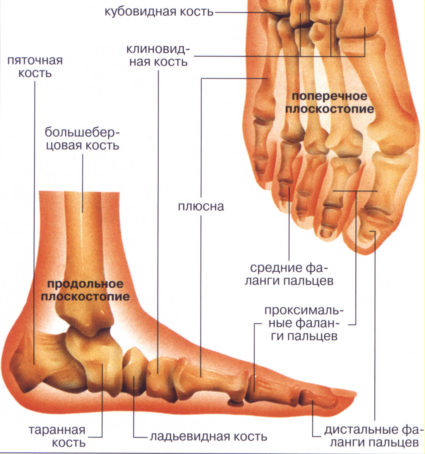
Одной из отличительных особенностей миодистрофии Дюшенна является сочетание данной формы с патологией костно-суставной системы и внутренних органов (сердечно-сосудистой и нейроэндокринной систем). Костно-суставные нарушения характеризуются деформациями позвоночника, стоп, грудины. На рентгенограммах обнаруживаются сужение костно-мозгового канала, истончение коркового слоя длинных диафизов трубчатых костей.
Сердечно-сосудистые расстройства клинически проявляются лабильностью пульса, артериального давления, иногда глухостью тонов и расширением границ сердца. На ЭКГ регистрируются изменения миокарда (блокада нож ек пучка Гиса и др.). Нейроэндокринные нарушения встречаются у 30–50 % больных. Чаще других наблюдаются синдром Иценко– Кушинга, адипозогенитальная дистрофия Бабинского–Фрелиха. Интеллект у многих больных снижен в различной степени.
Течение. Болезнь имеет быстро прогрессирующее злокачественное течение. К 7–10 годам возникают глубокие двигательные расстройства – выраженное изменение походки, снижение мышечной силы, в значительной степени ограничивающие свободное, самостоятельное передвижение больных. К 14–15 годам наступает обездвиженность.
Разновидности мышечной дистрофии
Выявлено более 30 видов мышечной дистрофии, из них наиболее распространены 9:
- миодистрофия Дюшенна – наиболее распространенная форма, развивается преимущественно у молодых мужчин;
- дистрофия Беккера – к сорокалетию больной теряет способность самостоятельно передвигаться;
- миотония – симптомы проявляются чаще у взрослых (либо у детей в период позднего детства);
- врожденная дистрофия – диагностируется сразу после рождения или в первые месяцы жизни;
- лимб-поясная – свойственна подросткам, молодым людям (20-25 лет), затрагивает мышцы бедер и плеч;
- плече-лопаточно-лицевая – вызывает ослабление мышц лица, плечевых мышц, теряется способность поднимать руки, возникают проблемы с речью, тахикардия;
- дистальная – для заболевания характерна слабость рук и нижних конечностей;
- дистрофия Эмери-Дрейфуса – для женщин опасна параличом сердца, у детей (преимущественно мальчиков), кроме сердца, поражаются икроножные мышцы и верхний плечевой пояс;
- болезнь Шарко (и прочие дегенеративные заболевания) также могут провоцировать схожие с мышечной дистрофией симптомы.
Из этих 9 форм мышечной дистрофии наиболее часто диагностируют следующие:
- Дистрофия Дюшенна. Симптомы проявляются в раннем детстве (до 5 лет). Болезнь диагностируют у одного из 3500 новорожденных мальчиков. Это наиболее распространенное смертельное генетическое расстройство. Женщины могут быть носителями гена, подвергшегося мутации, но сами этим расстройством не страдают. Дегенеративные изменения блокируют белок (дистрофин), поддерживающий структурные элементы мышечной ткани и клеточных мембран. Без дистрофина мышечная слабость прогрессирует, провоцируя обездвиженность и смерть.
- Дистрофия Беккера. Хотя производство дистрофина при этом заболевании сохраняется, уровень стержневидного белка существенно ниже нормы. Мышечная дистрофия развивается к 12-летнему возрасту. Болезнь развивается медленнее, но поражения значительны: искривление позвоночника, трудности с дыханием, постоянная усталость, слабость, болезни сердца, когнитивные проблемы.
Классификация миопатий
На сегодняшний день единой классификации миопатий не существует, эти заболевания подразделяют по нескольким принципам. Первой работой в этом направлении была так называемая клиническая классификация нервно-мышечных заболеваний, считающихся в те времена болезнями исключительно мышечных тканей. Согласно этой системе, медики выделяли такие типы миопатий, как конечностно-поясная, лице-плече-лопаточная, гумеро-тибиальная и другие.
Данное деление можно было считать условным, так как оно не отличалось четкостью характеристик, как и сегодня. Более точной классификации миопатий и нервно-мышечных заболеваний в медицине нет, поэтому до сих пор применяется существующая система.
Известна также патогенетическая классификация, появление которой связано с возникновением новых знаний о миопатии. К примеру, стало известно, что миодистрофии могут проявляться по причине множественного поражения нервов, которые могут спровоцировать нарушение обменных процессов и токсические воздействия. Так миопатии стали подразделяться на невральные амиотрофии, первично-мышечные заболевания и др.
Все детальнее с развитием медицины становится патогенетическая классификация, в основе которой лежит знание о пораженном заболеванием белке. Согласно этой классификации видами миопатии бывают кальпаинопатия, титинопатия и пр. Выявление дефективного белка позволяет сделать выводы о характере его мутации.
Миопатия подлежит разделению на наследственный и приобретённый тип. В анамнезе наследственных типов иногда содержатся относительно четкие данные о наличие этого заболевания у родственников. Примерами миопатий наследственного типа являются дистрофические миопатии, такие, как миопатия Дюшенна, митохондриальные заболевания, а также болезни накопления, наиболее распространена из которых болезнь Помпе.
Различные виды миопатий имеют разный патогенез, что напрямую зависит от того, какой ген в конкретном случае поражен. Так дистрофические миопатии или миодистрофии являются следствием процесса, в котором происходит нарушение синтеза структурных белков миофибрилл. Болезни накопления — результат снижения количества вырабатываемых ферментов и уменьшения их активности. В данном случае рано или поздно пациента начинает беспокоить мышечная слабость и атрофия мышц, что является ключевыми признаками миопатии.
Диагностика
Диагностика миодистрофии Дюшена ставится на основании следующих результатов осмотра и анализов:
- На ЭКГ выявляется поражение миокарда латеральной и задне-нижней стенок левого желудочка, что определяется по следующим показателям: высокий зубец наблюдается в отведении V6; глубокий зубец Q наблюдается в отведениях V6, aVF, 2 и 3.
- Также исследуется содержание дистрофина в мышечной ткани (при этом заболевании дистрофия не выявляется).
- В ходе биохимических исследований в плазме крови определяется активность КФК (фермента креатинфорсфокиназы), которая обычно существенно повышена (в том числе у носительниц гена). Иногда для уточнения источника исследуются изоферменты КФК.
- Проводится также генодиагностика.
- Фибрилляции на ЭМГ сообщают о некрозе мышечных волокон.
- Биопсия мышц является одним из основных методов диагностики миопатии Дюшена, причем выбирается умеренно пораженная мышца, поскольку очень ослабленная и существенно пострадавшая мышца окажется неинформативной.
Наиболее достоверными являются анализы на активность в сыворотке мышечных ферментов, биопсия мышц и ЭМГ (электромиография).
Симптомы миопатий
Основным симптомом, свидетельствующим о наличии одной из разновидностей миопатий, является постоянная слабость мышц. После отдыха это ощущение либо не проходит вовсе, либо становится чуть менее выраженным, но все равно весьма заметным.
Второй характерный признак — атрофия. Ткани становятся тонкими и крайне малоактивными. Атрофия может затрагивать только некоторые группы мышц, к примеру, отдел плечевого пояса или бедер или поражать все мышечные ткани в равной степени, полностью лишая человека возможности нормально двигаться.
Характерен для миопатии и крайне сниженный мышечный тонус. Каждая мышца в области поражения становится дряблой и вялой на ощупь. Соответственно, это сказывается на двигательной активности, которая не может оставаться на прежнем уровне.
Из-за того, что мышечный каркас перестает выполнять функцию поддержки туловища в правильном положении, позвоночник начинает испытывать чрезмерные нагрузки, в результате чего происходит его искривление. Позвоночник может изгибаться по-разному. Наиболее распространенные варианты — это искривление спереди назад, в результате чего развивается лордоз или кифоз, и вбок, как при обычном сколиозе. Искривление позвоночника при миопатии может принять такую серьезную степень, что уже само по себе будет являться значимой и сложно разрешимой проблемой.
Последний симптом миопатий — это появление псевдогипертрофии мышечных тканей. Из-за сильного истончения некоторых зон конечностей, мышцы других отделов внешне выглядят слишком крупными. Например, так происходит при атрофии мышц бедер, когда икры выглядят чрезмерно массивными на их фоне. На самом деле в данном случае как раз икроножные мышцы имеют нормальный размер, но визуально они выглядят неестественно увеличенными.
Причины генетического расстройства
Точно сказать, почему появляется врожденная миопатия у ребенка, не сможет ни один доктор. Факторы формирования этого нарушения в человеческом геноме не изучены, как и большинство причин других генетических отклонений.
Известно, что миопатия у детей начинает формироваться еще при внутриутробном развитии.
Важно! Уже в первые месяцы жизни можно заметить синдром у ребенка: он слишком вялый, пассивный, быстро утомляется и плохо двигается.

В течение первого полугода малыш испытывает острые затруднения при вставании на ножки, попытках сидеть, ему сложно держать головку. Практически всегда врожденная миопатия сопровождается болезнями внутренних органов:
- нарушается интеллектуальное развитие, появляются патологии умственной сферы, отсталость, психические отклонения, так как мозг и нервная система испытывают колоссальные нагрузки, не получают достаточного количества полезных веществ из-за врожденной структурной миопатии;
- появляются заболевания сердца и органов дыхания из-за общей слабости мышечного аппарата. Это самые опасные состояния, которые при врожденной миопатии нередко приводят к смерти;
- страдает костный мозг, из-за чего наблюдаются определенные изменения в развитии при врожденной структурной миопатии: деформируется позвоночник, у ребенка слишком высокое нёбо, обнаруживается врожденный вывих бедра.
Врачи полагают, что врожденные миопатии могут возникать из-за нарушений здоровья малыша, которые формируются еще до или после рождения в течение первых месяцев.
К этим нарушениям относят, прежде всего, родовые травмы, связанные с изменениями в структурах мышечных тканей. Также выделяют патологии соединительных структур, которые при врожденной миопатии очевидны уже с первых месяцев жизни.

Заболевание может зависеть от уровня креатина и креатинина в организме, замечена связь врожденной структурной миопатии с их показателями. Также врачи считают, что структурные нарушения могут быть последствием инфекций или неполучения малышом еще в период нахождения в утробе достаточного количества микроэлементов.
Что такое миопатия Дюшена
Это серьезное заболевание поражает, главным образом, мышцы в области туловища, бедер и плеч. При этом пациенты, как правило, могут свободно использовать руки и пальцы, но у них возникают проблемы с ходьбой, бегом, и так далее. Мышечная слабость прогрессирует постепенно. Обычно она проявляется в раннем детстве, но поначалу симптомы миопатии Дюшена выражены очень слабо. С возрастом они становятся все более выраженными, и приводят к резкому снижению качества жизни.
Миопатия Дюшена диагностируется приблизительно у одного из 3500 мальчиков.
В мышечной ткани содержится дистрофин – белок, необходимый для нормальной работы мышц. У людей с миопатией Дюшена этого вещества слишком мало. Со временем это приводит к повреждению мышечных волокон и ослаблению мышц. Причиной этого является особый ген, который передается от родителей к детям, либо генные мутации, произошедшие в период внутриутробного развития.
Для каждого сына женщины, которая является носителем гена Дюшена, вероятность развития миопатии Дюшена составляет ровно 50%. Дочери такой женщины станут носителями этого гена с такой же вероятностью.
Если у ребенка миопатия Дюшена, значит ли это, что у кого-то из членов семьи есть ген Дюшена? Не обязательно. Приблизительно в половине случаев заболевшие миопатией этого типа не получают дефектный ген от одного из родителей. В клетках плода еще во время беременности происходят мутации, результатом которых и становится миопатия. Это может произойти из-за «ошибки», которая случилась при копировании родительских генов в клетки, которые должны будут образовать организм ребенка. Почему это происходит, в настоящее время неизвестно.
Точно узнать, есть ли у кого-то из членов вашей семьи ген Дюшена, можно, только при помощи генетического консультирования.
Каковы симптомы?
Обычно первые симптомы миопатии Дюшена появляются в возрасте 1-3 лет. Родители могут заметить следующие признаки миопатии Дюшена:
- Ребенку трудно ходить, бегать, прыгать, подниматься по лестницам. Походка ребенка может отличаться от походки его ровесников – он ходит вразвалочку, и менее уверенно, чем остальные. Иногда дети с миопатией Дюшена начинают ходить позже остальных, однако и совершенно здоровые малыши иногда делают первые шаги несколько позже сверстников;
- В более старшем возрасте ребенок может опираться на руки, чтобы встать;
- У ребенка могут наблюдаться проблемы с обучением – как правило, не очень серьезные.
Иногда первым признаком миопатии Дюшена является замедленное развитие речи.
Как диагностируется
В первую очередь врачи, как правило, просто наблюдают за ребенком, в особенности за тем, как он ходит, бегает и встает с пола. Если основания подозревать миопатию Дюшена, будет назначен анализ крови на креатинкиназу – это фермент, уровень которого у людей с этим нарушением всегда очень высок (в 10-100 раз выше нормы). Если уровень креатинкеназы у ребенка в норме, миопатию Дюшена исключают и начинают искать другие причины появившихся у малыша симптомов.
Следующим этапом диагностики миопатии Дюшена является биопсия мышечной ткани и/или генетическое тестирование.
В ходе биопсии врач берет небольшой фрагмент мышечной ткани для дальнейших анализов; процедуру проводят под общей анестезией. Образец ткани изучают под микроскопом при помощи особых техник, чтобы оценить состояние мышечных волокон и количество дистрофина.
Для проведения генетического тестирования необходимо необходимое количество крови пациента. С помощью этого метода выявляют гены, которые отвечают за развитие миопатии Дюшена. В большинстве случаев этот способ позволяет точно диагностировать данное заболевание.
Что это такое
Патология названа в честь французского невролога, «отца электротерапии» Г. Бенджамена Армана Дюшена, который описал ее первым в 1853 г. По статистике, заболевает один из 4 тыс. мальчиков, поскольку миопатия передается по мужской линии.
Миодистрофия Дюшенна обусловлена повреждением генов, ответственных за синтез дистрофина. Роль этого мышечного белка трудно переоценить: он поглощает энергию, регулирует уровень кальция и контролирует рост мышц.
Если мышца сокращается, молекула дистрофина сжимается, как пружина. Благодаря «пружинящему» механизму мембраны миоцитов, соединительные ткани и сухожилия защищены от излишней механической нагрузки.
Миопатия Дюшенна передается аутосомно-рецессивно, то есть с перерывами, и зачастую наследуется через поколение. Около 25% случаев связаны с устойчивым изменением генетического кода в яйцеклетке матери. Остальные 75% объясняются ее гетерозиготностью.
В период генетического обследования ключевую роль играет выявление у сестер больного скрытых симптомов патологии. При их наличии существует 50%-вероятность передачи дефектного гена детям мужского пола, и половина дочерей станут его носительницами.
Женщины, у которых есть поврежденный ген, делятся им со своими детьми, при этом сами синдромом Дюшена не страдают. Ему подвержены преимущественно мальчики, но при повреждении структуры хромосом могут заболеть и девочки.
Диагностические мероприятия
Диагностика синдрома Дюшенна не вызывает трудностей у специалистов, поскольку имеет весьма специфические симптомы.
Врачи традиционно начинают с опроса больного или его родителей и сбора анамнестических данных
Особое внимание они уделяют:
- Времени появления первых симптомов,
- Локализации первичной мышечной слабости,
- Общему самочувствию пациента,
- Наличию подобных расстройств у родных и близких.
Во время неврологического обследования выявляется:
- Слабость определенной группы мышц и определяется ее степень,
- Изменение мышечного тонуса,
- Атрофические процессы,
- Гипо- и арефлексия,
- Деформация стопы, груди, позвоночного столба.
Врачи наблюдают за больным ребенком, обращая внимание на то, как он ходит, бегает и встает с пола. Изменение походки — важный диагностический признак миопатии
После проведения первичных диагностических процедур врачи могут заподозрить наличие у больного патологии и поставить предварительный диагноз. Чтобы его подтвердить или опровергнуть, пациента направляют на лабораторно-инструментальное обследование.
- Анализ крови на гормональный статус.
- Биохимический анализ крови на активность креатинкиназы – фермента, уровень которого у больных детей очень высок. Если КФК в норме, миопатию Дюшена исключают.
- Иммуногистохимическое исследование – микроскопия биоптата мышечной ткани, взятого от больного, с целью определения белка дистрофина. У лиц с миопатией он отсутствует.
- ДНК-тест – генетическое исследование крови больного, позволяющее определить мутантный ген и точно диагностировать патологию.
Дополнительные диагностические методики:
- Электрокардиография — выявление признаков поражения миокарда.
- Электромиография — определение фибрилляции, свидетельствующей о некрозе мышечных волокон. Эта методика оценивает состояние скелетной мускулатуры и подтверждает, что в основе патологии лежит именно поражение мышц, а не нарушение передачи нервных импульсов.
- Дыхательные пробы, рентгенография позвоночника и органов грудной клетки, УЗИ сердца — методы, не оказывающие существенного влияния на процесс постановки диагноза, но позволяющие выявить имеющиеся отклонения в структуре и функционировании органов дыхания и сердца.
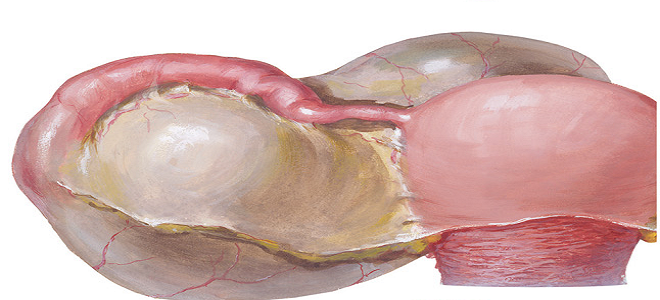
Киста Беккера — что это такое?
Другое название болезни – грыжа или бурсит подколенной ямки. Киста Беккера коленного сустава развивается в сумке, расположенной между сухожилиями полуперепончатой и икроножной мышц.
Она образуется при значительном скоплении синовиальной жидкости из-за травмы или длительно протекающего воспаления в суставных тканях. Киста ограничивает подвижность в колене, причиняет боль при движении.
Лучше понять, что это такое поможет общая схема: движения суставов обеспечивают эластичная хрящевая ткань, связочный аппарат и синовиальная жидкость. Последняя работает как смазка, уменьшая трение и циркулируя по всей суставной поверхности.
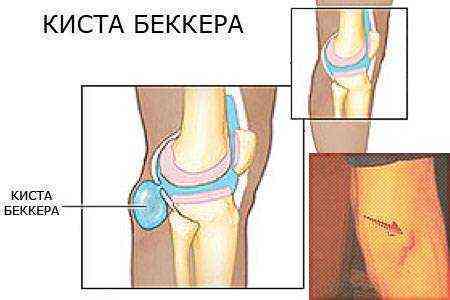
Киста Беккера под коленом, фото и схема
Определенное ее количество проникает и в область сухожилий. Если ее объем увеличивается, то она скапливается в межсухожильной сумке – так появляется киста Беккера.
Первичная причина образования – это патология в коленном суставе, например:
артрит, в частности ревматоидный; гонартроз; травма колена, сопровождаемая повреждением хряща; остеоартроз – почти в половине случаев приводит к патологии; воспаление менисков.
Воспалительный процесс сопровождается образованием патологического экссудата, который также проникает в межсухожильную подколенную сумку.
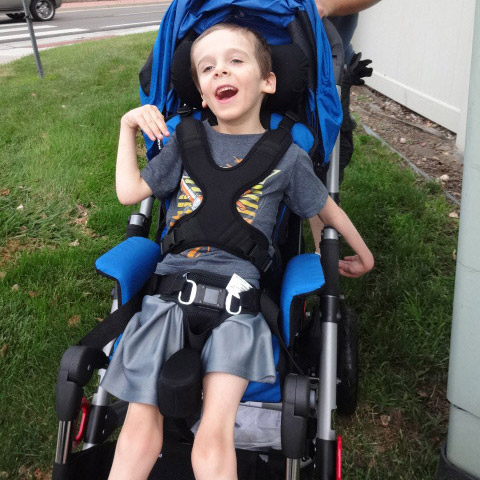
Управление на ранней стадии — когда ребенок гуляет, до 11 лет
- Физиотерапия для консультации по растяжке, для предотвращения контрактур.
- Позже ортезы на колено-стопу-лодыжку могут помочь продлить ходьбу.
- Серийное литье лодыжек может быть полезным (может предотвратить необходимость хирургического освобождения ахиллова сухожилия).
- Кортикостероиды:
- Они продлевают передвижение на 6-24 месяца. Они также могут помочь при дыхательной функции, кардиомиопатии и сколиозе.7
- Это должно быть сбалансировано с побочными эффектами, включая остеопороз и переломы позвонков.
- Преднизолон это обычное лечение. Есть рекомендуемые режимы дозирования.1
- Оптимизировать здоровье костей:1
- Витамин D и кальций диетические советы или добавки.
- Бисфосфонаты, если происходит перелом позвоночника.
Лечение миодистрофии Эрба-Рота
Этиопатогенетическая терапия пока не разработана. Симптоматическое лечение направлено на как можно более длительное сохранение двигательной способности пациента. С этой целью применяют медикаментозные курсы, включающие АТФ, витамины Е и группы В, тиоктовую кислоту и др. Занятия лечебной физкультурой должны проводиться ежедневно и включать упражнения на все группы мышц. Регулярно назначаются курсы массажа и физиопроцедуры.
При поражении сердечной мышцы рекомендован инозин, сердечные гликозиды, антиаритмики. При развитии контрактур может потребоваться ортопедическое лечение. Выраженное снижение жизненной емкости легких из-за атрофии дыхательных мышц служит показанием к ИВЛ.
Прогноз и профилактика
Мышечная дистрофия Эрба-Рота может иметь различную тяжесть и скорость прогрессирования, что бывает выражено даже в пределах одной семьи. Описаны тяжелые дюшенноподобные варианты заболевания с ранним летальным исходом от дыхательной недостаточности, инфекционных поражений легких или сердечной недостаточности. В относительно легких вариантах миодистрофия может протекать без поражения сердечной мышцы, обездвиженность больных наступает лишь к 50-летнему возрасту. Профилактикой является своевременное генетическое консультирование семейных пар, планирующих зачатие ребенка; исключение близкородственных браков, в которых оба супруга могут стать носителями патологического гена.
Признаки и симптомы мышечной дистрофии Дюшенна
Дети с мышечной дистрофией Дюшенна (МДД) часто поздно начинают ходить.

У малышей родители могут заметить увеличение икроножных мышц (см. изображение справа). Это увеличение известно как псевдогипертрофия, или «ложное увеличение» мышц, потому что мышечная ткань является ненормальной и может содержать рубцовую ткань.
Дошкольник с МДД может показаться неуклюжим и часто падать. Родители также могут заметить, что детям трудно подниматься по лестнице, вставать с постели или бегать.
К школьному возрасту детям тяжело ходить и передвигаться, ребенок часто падает. Чтобы сохранить равновесие, они могут выпячивать животы и откидывать плечи. Детям также трудно поднимать руки.
Многие дети с МДД начинают пользоваться инвалидной коляской в возрасте от 7 до 12 лет. Переход на инвалидную коляску обычно происходит постепенно; сначала кресло может потребоваться только для сохранения энергии ребенка при преодолении больших расстояний. (Дети часто вырабатывают новую самостоятельность после полного перехода на инвалидную коляску с электроприводом.)
В подростковом возрасте для действий, связанных с руками, ногами или туловищем, может потребоваться помощь или механическая поддержка.
Боль и чувствительность
Мышечная дистрофия Дюшенна само по себе обычно не болезненна. Некоторые люди иногда сообщают о мышечных судорогах; обычно их можно лечить безрецептурными болеутоляющими средствами.
Поскольку мышечная дистрофия не влияет непосредственно на нервы, осязание и другие чувства остаются нормальными, как и контроль над гладкими или непроизвольными мышцами мочевого пузыря, кишечника и половыми функциями.
Сердце
Недостаток дистрофина может ослабить мышечный слой сердца (миокард), что приводит к состоянию, которое называется кардиомиопатией. Со временем, иногда еще в подростковом возрасте, ущерб, нанесенный МДД сердцу, может стать опасным для жизни. Сердце должно тщательно и постоянно контролироваться, как правило, детским кардиологом.
Дыхательная функция
Начиная примерно с 10-летнего возраста, диафрагма и другие мышцы, управляющие легкими, могут ослабнуть, что сделает легкие менее эффективными при движении воздуха внутрь и наружу. Хотя ребенок может не жаловаться на одышку, к числу проблем и признаков, указывающих на плохое состояние дыхания, относятся головные боли, умственная отсталость, трудности с концентрацией или бессонницей, а также ночные кошмары.
Ослабленные дыхательные мышцы затрудняют кашель, что приводит к повышенному риску серьезной респираторной инфекции. Простая простуда может быстро перерасти в пневмонию
Важно сделать прививку от гриппа, а при возникновении инфекции — получить быстрое лечение
Неспособностью к обучению
Около треть мальчиков с МДД имеют некоторую степень неспособности к обучению, хотя немногие имеют серьезную умственную отсталость.
Врачи считают, что аномалии дистрофина в мозге могут оказывать незначительное влияние на когнитивные функции и поведение. Проблемы с обучением при МДД возникают в трех основных областях:
- концентрация внимания;
- словесное обучение и память;
- эмоциональное взаимодействие.
Дети с подозрением на нарушение способности к обучению могут быть обследованы психоневрологом-воспитателем или педиатром.
Если диагностирована неспособность к обучению, образовательные и психологические вмешательства могут начаться сразу же. Специалист может назначить упражнения и методы, которые могут помочь улучшить эти области, а специализированные школы могут оказать особую помощь в обучении.
Классификация миопатий
С учетом того, какие мышцы подвержены повреждениям, преобразуется классификация болезни. Миопатия Дюшена считается наиболее распространенной разновидностью и отличается наивысшей степенью сложности. Эта патология имеет наследственный характер в большинстве примеров, развивается стремительно. Проблема с работой дистрофина, регулирующего прочность мембран, является основной причиной возникновения болезни.
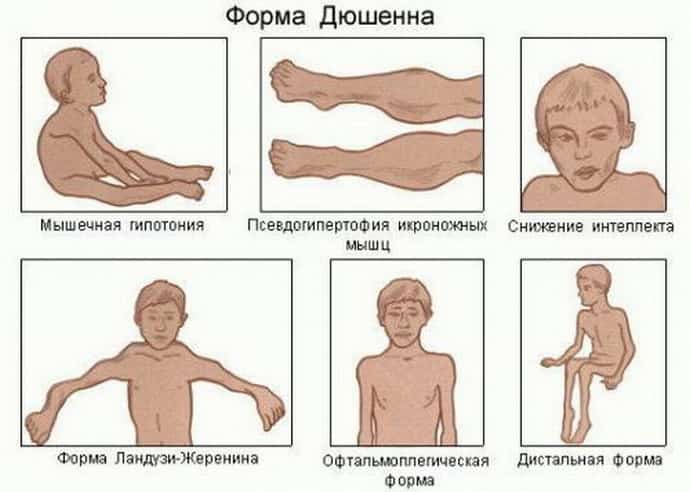
Зачастую такая патология возникает только у мужчин. Девушки в большинстве примеров выступают в роли переносчиков. Развивается такая миопатия с трехлетнего возраста, отличается отчетливой симптоматикой, о которой свидетельствует заболевание.
При миопатии Беккера возникает сердечная недостаточность. Это усугубляет общее положение. Преобразование можно проследить на первичных этапах при повреждении миокарда. Это все можно определить после выполнения ЭКГ или ЭхоКГ. Если пренебречь терапией этой патологии, может развиваться слабость в мышцах и проблемы с дыханием. Такая симптоматика может привести к летальному исходу.
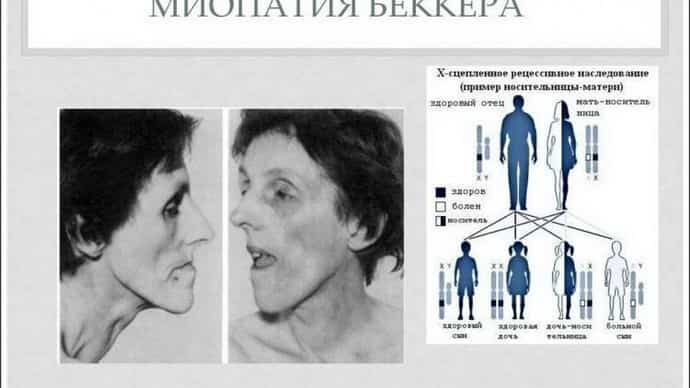
Для миопатии Беккера различают 2 способа диагностики:
- Генодиагностика.
- Анализ дистрофина в мышечных тканях.
Зачастую ко второму варианту прибегают при подозрениях на эту болезнь, поэтому диагноз может быть подтвержден или опровергнут.
Главная терапевтическая методика направлена на предотвращение патологии. ЛФК может поспособствовать уменьшению мышечных преобразований, используются приспособления, облегчающие передвижение. К хирургическим процедурам прибегают в сложных ситуациях. Миопатия при этом угрожает жизнедеятельности людей.
Главной причиной заболевания является генетическое преобразование, развивающееся в процессе внутриутробного развития. Миопатия Эрба может передаваться по наследству. Наиболее отчетливым признаком является слабость м мышечных тканях. Наблюдается атрофирование и прекращение их возможного развития.
Поэтому через какое-то время они высыхат, исчезает возможность свободно передвигаться. При этом пациенты не испытывают болезненные симптомы, присутствует только слабость. Она не исчезает даже после продолжительного сна, через какое-то время начинает усугубляться.

Дальнейшее чтение и ссылки
-
Мышечная дистрофия, тип Дюшенна, МДД; Онлайн менделевское наследство в человеке (OMIM)
-
Аталурен для лечения мышечной дистрофии Дюшенна с бессмысленной мутацией в гене дистрофина; Руководство по высокоспециализированным технологиям NICE, июль 2016 г.
-
Манзур А.Ю., Кинали М, Мунтони Ф; Обновленная информация по управлению мышечной дистрофии Дюшенна. Arch Dis Child. 2008 Nov93 (11): 986-90. Epub 2008 30 июля.
-
Бейтиа Мде Л, Врай Дж, Киршнер Й; Медикаментозное лечение мышечной дистрофии Дюшенна: имеющиеся данные и перспективы. Acta Myol. 2012 май 31 (1): 4-8.
-
Бушби К, Финкель Р, Бирнкрант DJ и др.; Диагностика и лечение мышечной дистрофии Дюшенна, часть 1: диагностика, фармакологическое и психосоциальное лечение. Ланцет Нейрол. 2010 янв. 9 (1): 77-93. doi: 10.1016 / S1474-4422 (09) 70271-6. Epub 2009 27 ноября.
-
Леунг Д.Г., Вагнер К.Р.; Терапевтические достижения при мышечной дистрофии. Энн Нейрол. 2013 Sep74 (3): 404-11. doi: 10.1002 / ana.23989.
-
Angelini C, Tasca E; Усталость при мышечных дистрофиях. Нервно-мышечный диссонанс. 2012 г., 22 декабря, добавление 3: S214-20. doi: 10.1016 / j.nmd.2012.10.010.
-
Бушби К, Финкель Р, Бирнкрант DJ и др.; Диагностика и лечение мышечной дистрофии Дюшенна, часть 2: осуществление междисциплинарной помощи. Ланцет Нейрол. 2010 Feb9 (2): 177-89. doi: 10.1016 / S1474-4422 (09) 70272-8. Epub 2009 27 ноября.
-
Манзур А., Кунцер Т., Пайк М. и др.; Глюкокортикоидные кортикостероиды для мышечной дистрофии Дюшенна. Кокрановская база данных Syst Rev. 2008 янв. 23 (1): CD003725.
-
Чеук Д.К., Вонг В., Рэйдж Э. и др.; Операция при сколиозе при мышечной дистрофии Дюшенна. Кокрановская база данных Syst Rev. 2015, октябрь 110: CD005375. doi: 10.1002 / 14651858.CD005375.pub4.
-
Респираторная помощь пациенту с мышечной дистрофией Дюшенна — ATS Consensus Statement 2004; Американское торакальное общество
-
Birnkrant DJ, Panitch HB, Benditt JO и др.; Консенсусное заявление Американского колледжа грудных врачей о респираторном и соответствующем лечении пациентов с мышечной дистрофией Дюшенна, подвергающихся анестезии или седации. Грудь. 2007 Dec132 (6): 1977-86.
-
Гурнаней Х, Браун А, Литман Р.С.; Злокачественная гипертермия и мышечные дистрофии. Anesth Analg. 2009 Октябрь 2009 (4): 1043-8. doi: 10.1213 / ane.0b013e3181aa5cf6.
-
Сенкевич Д., Кулак В., Окуровска-Завада Б. и др.; Мышечная дистрофия Дюшенна: современная клеточная терапия. Ther Adv Adv Neurol Disord. 2015 Jul8 (4): 166-77. doi: 10.1177 / 1756285615586123.
-
Штрехле Э.М., Штрауб V; Последние достижения в лечении мышечной дистрофии Дюшенна. Arch Dis Child. 2015 дек100 (12): 1173-7. doi: 10.1136 / archdischild-2014-307962. Epub 2015 7 июля.
-
Goyenvalle A, Seto JT, Davies KE и др.; Терапевтические подходы к мышечной дистрофии. Хум Мол Генет. 2011 апр 1520 (R1): R69-78. дои: 10.1093 / мг / ддр105. Epub 2011 24 марта.
-
Конечный П, Свидерски К, Чемберлен Ю.С.; Генная и клеточная терапия мышечной дистрофии. Мышечный нерв. 2013 май47 (5): 649-63. дои: 10,1002 / мус.23738. Epub 2013 29 марта.
-
Мерегалли М., Фарини А., Коллеони Ф. и др.; Роль стволовых клеток в мышечных дистрофиях. Curr Gene Ther. 2012 июнь 12 (3): 192-205.
-
Romfh A, McNally EM; Оценка состояния сердца при мышечной дистрофии Дюшенна и Беккера. Curr Heart Fail Rep. 2010 Dec7 (4): 212-8. doi: 10.1007 / s11897-010-0028-2.
-
Руководство по респираторному лечению детей с нервно-мышечной слабостью; Британское торакальное общество (2012)
-
Авторы не указаны; Наблюдение за состоянием сердечно-сосудистой системы у лиц, страдающих мышечной дистрофией Дюшенна или Беккера. Педиатрия. 2005 Dec116 (6): 1569-73.
-
Маврогени С., Маркунис-Маврогенис Г., Папавасилиу А. и др.; Поражение сердца при мышечной дистрофии Дюшенна и Беккера. Мир Дж Кардиол. 2015 июл 267 (7): 410-4. doi: 10.4330 / wjc.v7.i7.410.
-
Fairclough RJ, Бареджа А, Дэвис К.Е.; Прогресс в терапии мышечной дистрофии Дюшенна. Exp Physiol. 2011 Nov96 (11): 1101-13. doi: 10.1113 / expphysiol.2010.053025. Epub 2011 июл 31.
-
Desguerre I, Christov C, Mayer M и др.; Клиническая гетерогенность мышечной дистрофии Дюшенна (МДД): определение субфенотипов и прогностических критериев при длительном наблюдении. УТВЕРЖДАЕТ. 20094 (2): e4347. doi: 10.1371 / journal.pone.0004347. Epub 2009 5 февраля.
-
Холлоуэй С.М., Уилкокс Д.Е., Уилкокс А. и др.; Ожидаемая продолжительность жизни и смерть от кардиомиопатии среди носителей мышечной дистрофии Дюшенна и Беккера в Шотландии. Сердце. 11 октября 2007 г.
-
Манзур А.Ю., Мунтони Ф; Диагностика и новые методы лечения мышечной дистрофии. J Neurol Neurosurg Психиатрия. 2009 Jul80 (7): 706-14.
Профилактика и прогноз
Супругам, в роду которых имелись случаи наследственных заболеваний, перед планированием беременности необходимо посетить врача-генетика. Профилактика патологии также заключается в проведении пренатальной диагностики. Выявив миопатию на ранних сроках, можно прервать беременность.
Миопатия Дюшена — наследственная патология, отличающаяся тяжелым течением и быстрым прогрессированием. Это современная медицинская проблема, характеризующаяся разрушением мышечной ткани и быстрым развитием мышечной слабости. Все без исключения больные погибают в раннем возрасте из-за развития не совместимых с жизнью осложнений. Только адекватная и комплексная терапия, четкое соблюдение рекомендаций врача, тщательный уход и забота родителей могут замедлить ход болезни.
Больные быстро становятся инвалидами и погибают в совсем юные годы. Самое страшное, что детям с миопатией не в силах помочь даже квалифицированные врачи, современные медицинские технологии и терапевтические методики 21 века. Болезнь до сих пор остается неизлечимой, забирая молодые жизни. Современные ученые-медики всего мира трудятся над созданием радикального способа борьбы с миопатией Дюшенна.