Фенилкетонурия (фку). причины, симптомы, диагностика и лечение фенилкетонурии. лечение фенилкетонурии: диета и питание
Содержание:
- Прогноз и профилактика фенилкетонурии
- Диагностика
- Стандартные методы лечения
- Симптомы фенилкетонурии
- Диагностика
- Ответы на часто задаваемые вопросы
- Особенности питания новорожденных и диетотерапия
- Симптомы и диагностика патологии
- Клиническая картина
- Лечение фенилкетонурии
- Симптомы
- Общее описание
- Лечение
- Профилактика
Прогноз и профилактика фенилкетонурии
Проведения массового скрининга на фенилкетонурию в неонатальном периоде позволяет организовать раннюю диетотерапию и предотвратить тяжелые церебральные повреждения, нарушения функции печени. При раннем назначении элиминационной диеты при классической фенилкетонурии прогноз развития детей хороший. При поздно начатом лечении прогноз в отношении умственного развития неблагоприятный.
Профилактика осложнений фенилкетонурии заключается в проведении массового скрининга новорожденных, раннего назначения и длительного соблюдения диетического питания.
С целью оценки риска рождения ребенка с фенилкетонурией предварительное генетическое консультирование должны пройти супружеские пары, уже имеющие больного ребенка, состоящие в кровнородственном браке, имеющие родственников с данным заболеванием. Женщины с фенилкетонурией, планирующие беременность, должны соблюдать строгую диету до зачатия и во время беременности для исключения повышения уровня фенилаланина и его метаболитов и нарушения развития генетически здорового плода. Риск рождения ребенка с фенилкетонурией у родителей-носителей дефектного гена, составляет 1:4.
Фенилкетонурия (ФКУ)
– довольно редкое наследственное заболевание, связанное с нарушением обмена аминокислот. Организм больного фенилкетонурией человека не способен расщеплять аминокислоту фенилаланин
, которая поступает с белковой пищей. В результате этого, в тканях накапливаются соединения, отравляющие нервную систему и головной мозг в частности. Развивается умственная отсталость (малоумие), вплоть до идиотии. В связи с этим болезнь получила и другое название – фенилпировиноградная олигофрения.
Однако из всех наследственных заболеваний фенилкетонурия, единственное, которое удается полностью нейтрализовать. Сегодня ребенка, рожденного с признаками ФКУ, можно вырастить абсолютно здоровым. Обезопасить мозг малыша удается с помощью специальной диеты, о которой мы расскажем ниже.
В разных странах частота этого заболевания отличается в разы. В России рождается один больной ребенок на 10 000. В некоторых регионах Великобритании этот показатель в два раза выше – 1:5000. Дети на Африканском континенте практически не болеют фенилкетонурией. Среди больных количество девочек почти в два раза превышает количество мальчиков.
Диагностика
 Скрининговые тесты на содержание фенилаланина всем новорожденным проводят в роддоме.
Скрининговые тесты на содержание фенилаланина всем новорожденным проводят в роддоме.
В связи с тем, что фенилкетонурия сопровождается развитием необратимых умственных нарушений, во многих странах мира, в том числе и в России, принято использовать скриниг-методы диагностики. Что это означает? Всем без исключения новорожденным детям в роддоме проводят экспресс-тесты на содержание фенилаланина. Для этого берут капиллярную кровь (из пятки) на 4-5-й день жизни ребенка (у недоношенных на 7-й), наносят на специальный бумажный бланк и отправляют в лабораторию, где по определенным изменениям врач-лаборант делает выводы о содержании фенилаланина в крови. Отрицательный тест говорит об отсутствии фенилкетонурии.
Если тест оказывается положительным, то тогда проводят дополнительные исследования для определения содержания фенилаланина в крови и моче (хроматографию, флюориметрию). Концентрацию фенилаланина в крови и моче регулярно проверяют при проведении лечения, чтобы контролировать эффективность диеты и корригировать ее при необходимости.
Возможно проведение генетического исследования для подтверждения мутации в гене, отвечающем за фенилаланин-4-гидроксилазу. Подобное исследование возможно в качестве пренатальной диагностики, то есть на этапе беременности (берут околоплодные воды путем пункции). Это инвазивное исследование делают по строгим показаниям (например, наличие больного фенилкетонурией ребенка в семье). Выявление генетического дефекта у плода позволяет прервать беременность.
Стандартные методы лечения
Целью лечения фенилкетонурии является поддержание уровня фенилаланина в плазме в пределах 120–360 мкмоль/л (2–6 мг/дл). Обычно это достигается за счет тщательно спланированной и контролируемой диеты
Следует осторожно ограничивать потребление фенилаланином ребенком, поскольку это незаменимая аминокислота. Тщательно соблюдаемая диета может предотвратить умственную отсталость, а также неврологические, поведенческие и дерматологические проблемы
Лечение следует начинать в очень молодом возрасте, в противном случае можно ожидать некоторой степени умственной отсталости. Однако даже некоторые дети, получившие позднее лечение, неплохо справились. Исследования неоднократно демонстрировали, что дети с фенилкетонурией, которых лечат питанием с низким содержанием фенилаланина в возрасте до трех месяцев, чувствуют себя хорошо, их коэффициент интеллекта в пределах нормы.
Если люди с фенилкетонурией перестают контролировать потребление фенилаланина с пищей, обычно возникают неврологические изменения. Коэффициент интеллекта может снизиться. Другие проблемы, которые могут появиться и стать серьезными после прекращения диеты, включают трудности в школе, поведенческие проблемы, изменения настроения, плохую зрительно-моторную координацию, плохую память, плохие навыки решения проблем, усталость, тремор, плохую концентрацию и депрессию.
После долгих лет споров среди клиницистов сейчас почти все согласны с тем, что диету нужно продолжать бесконечно и что взрослые с фенилкетонурией, которые прекратили диету в детстве или позже, должны вернуться к ней. Многие молодые люди возобновили диету и обнаружили улучшение ясности ума в результате снижения уровня фенилаланина в крови.
Поскольку фенилаланин содержится практически во всех натуральных белках, невозможно адекватно ограничить рацион, используя одни только натуральные продукты, без ущерба для здоровья. По этой причине полезны специальные пищевые продукты, не содержащие фенилаланина. Продукты с высоким содержанием белка, такие как мясо, молоко, рыба и сыр, как правило, не допускаются в рацион. Продукты с низким содержанием белка, такие как фрукты, овощи и некоторые злаки, разрешены в ограниченных количествах.
В 2007 году Куван (гидрохлорид сапроптерина) был одобрен управлением по контролю за продуктами и лекарствами США (FDA) для лечения фенилкетонурии. Куван — это пероральный фармацевтический препарат BH4, природного кофактора фермента ФАГ (англ. PAH), который стимулирует активность остаточного фермента PAH по метаболизму фенилаланина в тирозин. Куван следует использовать в сочетании с диетой с ограничением фенилаланина. Куван производится компанией BioMarin Pharmaceutical Inc.
В 2018 году Палинзик (пегвалиаза) был одобрен FDA для взрослых с ФКУ. Палинзик — это инъекционная ферментная терапия для пациентов, у которых на текущем лечении наблюдается неконтролируемая концентрация фенилаланина в крови. Палинзик также производится компанией BioMarin Pharmaceutical Inc.
Симптомы фенилкетонурии
Симптомы заболевания у детей с классической формой патологии сразу после рождения не наблюдается. Однако такие дети обладают рядом специфических внешних признаков. У них имеет место быть:
- суховатая кожа белого оттенка;
- пигментация отсутствует практически полностью;
- волосы светлого оттенка;
- голубые глаза.
В возрасте 2 — 6 месяцев начинают проявляться симптомы заболевания. К вышеуказанным признакам добавляется вялость, пассивное восприятие окружающего мира, повышенная раздражительность и задержка психомоторного развития. Иногда возможно возникновение частой рвоты. Появляется беспричинное беспокойство. Могут наблюдаться приступы судорог. Если патологий не выявлено, в рацион ребенка вводят белковую пищу, и симптоматика начинается нарастать. Череп такого ребенка несколько уменьшен в размерах. Дети, страдающие патологией, начинают ходить позже сверстников. В год такие пациенты не могут выразить голосом эмоции. Они не воспринимают речь взрослых. Возможна задержка роста.
Не преобразованный фенилаланин выходит с потом и мочой, что приводит к появлению специфического затхлого запаха. Патология проявляется также в своеобразных позах и походке. Они возникают из-за того, что мышечный тонус больного повышен. В положении стоя ребенок широко расставляет ноги и сгибает их в коленях и тазобедренных суставах. При этом голова и плечи опущены. Походка шатающаяся. Ребёнок делает мелкие шаги. Больные сидят в так называемой позе портного. Они поджимают и скрещивают ноги. После 3 лет наблюдается повышенная возбудимость ребенка и его быстрая утомляемость. Присутствует нарушение поведения и психические расстройства. Наблюдается умственная отсталость. Очень часто болезнь сопровождает экзема, дерматит и аритмия. Если лечение отсутствует, состояние больного ухудшится. Своевременная постановка диагноза позволит уменьшить нарушения, с которыми может столкнуться ребёнок.
Диагностика
Фото: aidsmap.com
Во всех родильных домах проводится массовое тестирование детей на наличие повышенного содержание фенилаланина в крови. Исследование проводится на четвёртый день жизни. Материалом для тестирования служит кровь, взятая из пятки новорождённого. Далее из родильного дома биологический материал доставляют в медико-генетическую консультацию, где проводят полное исследование крови на содержание фенилаланина в крови. Используются различные подходы к выявлению заболевания:
- Тест Гатри – в его основе лежит ингибирование бактерий Bacillus subtilis. Действие ингибитора заканчивается при повышенном содержании фенилаланина в крови. Следовательно колонии бактерий начинают размножаться. Для оценки проведённого теста его сравнивают с определёнными стандартными значениями. Чувствительность теста составляет более 99%. Этот метод применяется во многих странах мира как скрининговый метод выявления фенилкетонурии. В Российской Федерации массово данный тест не применяется.
- Хроматография – метод основывается на движении частиц (молекул белков, аминокислот) в электрическом поле и последующем естественном разделении на однородные фракции. Соответственно образуется фракция фенилаланина в определённом месте в сорбенте. Для оценки теста его сравнивают со стандартными показателями. В качестве скринингово метода не применяется.
- Флюриметрия – определение количества фенилаланина в крови методом хроматографии с помощью автоматических флюориметров. Этот метод во многих странах мира применяют как основной метод автоматизированного скрининга фенилкетонурии.
- Тандемная масс-спектрометрия – метод позволяет установить не только количество фенилаланина в крови, но и соотношение фенилаланин/тирозин.
Далее для уточнения диагноза применяют молекулярно-генетические методы диагностики. Они позволяют установить конкретную поломку гена и ,соответственно, дисфункцию конкретного фермента. Это может оказаться решающим моментом в лечении фенилкетонурии.
Ответы на часто задаваемые вопросы
Как проявляется фенилкетонурия у новорожденных?
Как выглядят больные фенилкетонурией?
- посветление волос и радужки глаза из-за недостатка пигмента меланина
- чрезмерная прибавка в весе
- быстро зарастает большой родничок
- суховатая кожа
- шелушение, сыпь и экзема
- частая рвота
- моча и пот с характерным «мышиным» запахом
- появляются судороги и спазмы
- скованность движений и зажатая «поза портного», что связанно с повышенным напряжением в мышцах
- неадекватное поведение, выкрики, смех
- уменьшение размеров черепа
- деформация ушных раковин
- дрожание пальцев рук
- недержание мочи
- выступающая вперед нижняя челюсть
Какие смеси использовать для ребенка с фенилкетонурией?
витаминымикроэлементыДля детей до одного года рекомендуют:
- Афенилак 13, Афенилак 15 от компании «Нутритек», Россия;
- MIDмил ФКУ 0 (Hero, Испания);
- ХР Аналог («Нутриция», Голландия);
- Фенил Фри 1 («Мид Джонсон» США).
Для детей старше одного года и для взрослых:
- П-АМ 1, П-АМ 2, П-АМ 3;
- Изифен (готовый продукт), а также ХР Максамейд и ХР Максамум с нейтральным и фруктовым вкусами («Нутриция», Голландия).
Какие бывают типы фенилкетонурии?
фенилкетонурии
- Фенилкетонурия I. Классическая и наиболее распространенная форма заболевания, описанная выше в статье. Связана с мутацией гена в 12-й хромосоме, при этом нарушается образование фермента фенилаланин-4-гидроксилазы, который превращает фенилаланин в тирозин.
- Фенилкетонурия II. При этой форме заболевания нарушение происходит в 4-й хромосоме. Нарушается выработка фермента дигидроптеридинредуктазы, который также способствует превращению фенилаланина в тирозин. Заболевание наследуется так же, как и I форма: для того, чтобы родился больной ребенок, необходимо, чтобы носителями гена были оба родителя. Распространенность фенилкетонурии II – 1 случай на 100 000 новорожденных.
- Фенилкетонурия III. В результате генетических нарушений возникает недостаток фермента 6-пирувоилтетрагидроптеринсинтазы. Наследуется, как и две предыдущие формы заболевания. Распространенность – 1 случай на 300 000 новорожденных.
Дают ли инвалидность при фенилкетонурии?
Критерии установления инвалидности при фенилкетонурии
- При фенилкетонурии I инвалидность устанавливают только при необратимых нарушениях со стороны центральной нервной системы, которые приводят к неврологическим расстройствам и умственной отсталости.
- При фенилкетонурии II и III типа группу инвалидности устанавливают во всех случаях.
Существует ли профилактика фенилкетонурии?
- Генетическое консультирование. Необходимо людям, планирующим завести ребенка, которые больны или являются носителями неправильного гена, у которых болен хотя бы один близкий родственник или уже родился больной ребенок. Консультирование проводит врач-генетик. Он помогает разобраться, как ген, ответственный за фенилкетонурию, передавался в предыдущих поколениях, каковы риски будущего ребенка. Также генетик помогает с планированием семьи.
- Скрининг новорожденных. Анализ не помогает предотвратить заболевание, но позволяет выявить его максимально рано, пока оно еще не привело к необратимым изменениям в головном мозге.
- Консультации и диета для женщин, страдающих фенилкетонурией. Если вы женщина и страдаете ФКУ, вам следует проконсультироваться с врачом и спросить, когда лучше планировать беременность в вашем случае. Во время беременности нужно соблюдать правильную диету – это помогает предотвратить дефекты развития у ребенка.
Каковы факторы риска фенилкетонурии?
- Как уже упоминалось в статье, ребенок рискует получить заболевание или стать носителем мутантного гена, если он есть у обоих родителей.
- Среди разных этнических групп распространенность фенилкетонурии различается. Например, среди представителей негроидной расы неправильный ген встречается реже.
- В группе повышенного риска находятся дети матерей, страдающих фенилкетонурией. Если во время беременности женщина не придерживается специальной диеты, у ребенка могут возникать дефекты развития.
Особенности питания новорожденных и диетотерапия
На первом году жизни ребенка с ФКУ допустимо кормление грудным молоком, но его количество должно быть ограничено. До 6 месяцев допустимым уровнем потребления фенилаланина является 60-90 мг на 1 кг веса малыша (в 100 г молока содержится 5,6 мг фенилаланина). Начиная с 3 месяцев, рацион ребенка следует поэтапно расширять, вводя в него фруктовые соки и пюре.
Детям с 6 месяцев разрешено введение в рацион овощных пюре, каш (из саго), безбелковых киселей. После 7 месяцев можно давать малышу низкобелковые макаронные изделия, с 8 месяцев – хлеб, не содержащий белка. Возраст, до которого следует ограничивать поступление белка в организм больного ребенка, не установлен. Врачи до настоящего времени дискутируют по вопросу целесообразности пожизненной диетотерапии, но сходятся во мнении, что минимум до 18 лет необходимо придерживаться диетического питания.
Фенилкетонурия, диагностированная у женщины, не является поводом отказаться от рождения ребенка. Будущим мамам с ФКУ для предупреждения повреждения плода во время беременности и профилактики возможных осложнений необходимо до планируемого зачатия и во время вынашивания ребенка придерживаться диеты с ограничением фенилаланина (его уровень в крови должен быть до 242 мкмоль/л).
Безлактозные смеси для малышей
Диета при фенилкетонурии базируется на существенном уменьшении дозы натурального белка в суточном рационе, но организм новорожденного ребенка не может развиваться нормально при отсутствии необходимых микроэлементов. Для восполнения потребности малыша в белке применяются безлактозные аминокислотные смеси, которыми, согласно российскому законодательству, больные должны обеспечиваться бесплатно.
Толерантность грудничка к фенилаланину в течение первого года жизни стремительно изменяется, поэтому необходимо контролировать его концентрацию в крови ребенка и вносить корректировки в диету. Смеси рассчитаны на определенные возрастные группы:
- малышам до года назначаются Афенилак 15, Аналог-СП, PKU-1, PKU-mix, PKU Anamix;
- детям, старше 1 года, назначают обогащенные витаминами и минералами смеси с повышенным содержанием белка – PKU Prima, P-AM Universal, ФКУ-1, ФКУ-2, ХР Максамейд, ХР Максамум.

Диетические продукты для пополнения запасов белка
Одним из основных компонентов пищевой диеты при фенилкетонурии являются малобелковые продукты на основе крахмала. Эти добавки содержат гидролизат казеина, триптофан, тирозин, метионин, азот и обеспечивают суточную потребность организма ребенка в белке, который необходим для нормального развития и роста. Специализированными продуктами, восполняющими нехватку необходимых минералов и аминокислот при их нехватке в рационе питания, являются:
- Берлофен;
- Циморган;
- Минафен;
- Апонти.
Симптомы и диагностика патологии
При фенилкетонурии симптомы можно заметить со 2 месяца жизни ребенка. Получая белок с молоком или его заменителями, организм сталкивается с первыми проблемами метаболизма. Это проявляется вялостью или, наоборот, гипервозбудимостью, частыми срыгиваниями и рвотой, иногда дистонией мышц и даже судорогами.
После полугода становятся видны некоторые задержки психомоторного развития: ребенок теряет активность, не пытается подниматься, садиться, стоять; может не узнавать мать и других родных, не реагировать на визуальные раздражители (яркие, цветные игрушки).
Начинает шелушиться кожа, иногда доходит до дерматитов и экземы, что обусловлено ненормальным составом пота. Моча имеет выраженный плесневый запах из-за избытка фенилпировиноградной, фенилуксусной и фенилмолочной кислот.
По мере роста ребенка начинают проявляться и другие признаки. Среди физических симптомов отмечают меньший по отношению к нормальным показателям размер головы. Прорезывание зубов начинается поздно — часто после года, при этом высока вероятность гипоплазии эмали. При попытках стоять ноги широко расставлены, при этом согнуты в коленях, также опущены плечи и голова. Это происходит из-за гипертонуса мышц.
В 2-3 года проявляется явное нарушение речи: задержка речевого развития по сравнению с ровесниками или полная неспособность говорить. К 4 годам вероятнее всего диагностируется олигофрения.
Раннее обнаружение данного заболевания, как и ряд других болезней, включено в скрининговые программы, поэтому в роддомах проверяют всех родившихся детей. Для диагностики ФКУ анализ крови и мочи берется на 2-3 день у доношенного младенца и на 7 — у тех, кто родился раньше срока. При выявлении в крови повышенного уровня фенилаланина, а в моче — его производных, характерных для данной болезни, назначаются подтверждающие исследования: биохимия мочи, анализ активности ферментов печени, дополнительно — консультация невролога, энцефалограмма и МРТ мозга.
Клиническая картина
Для больных фенилкетонурией характерен светлый цвет волос и голубая радужная оболочка глаз (дети всегда светлее своих родителей и здоровых братьев и сестер). В первые 2_3 месяца жизни большинство детей выглядит совершенно здоровыми. Лишь у некоторых из них отмечается вялость, сонливость, реже беспокойство. Ранним симптомом является рвота. Отмечается своеобразный запах мочи ребенка (затхлый, мышиный). К концу первого полугодия жизни у 20—50% больных детей появляются экзематозные изменения кожи, иногда значительные, у некоторых развивается склередема (см. Склередема новорожденных). В этом же возрасте начинает выявляться задержка психического развития, к-рая, нарастая, в дальнейшем приводит к тяжелому слабоумию (см.) — имбецильности или идиотии. Лишь у незначительного числа больных поражение интеллектуальной сферы выражено нерезко. Отмечается отставание в физическом развитии. Задерживается формирование двигательных навыков. У многих больных появляются судороги, которые могут постепенно исчезать даже без лечения, обнаруживается гилеррефлексия, непроизвольные движения. У большинства больных на ЭЭГ регистрируется пароксизмальная активность. У некоторых детей отмечается спазм мышц конечностей (преимущественно ног). Все отмеченные изменения характерны для больных, не получающих с рождения патогенетического лечения.
Лечение фенилкетонурии
Главным способом лечения фенилкетонурии является диетотерапия, ограничивающая поступление в организм белка и фенилаланина.
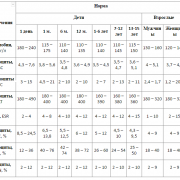
Основным критерием адекватности диеты при фенилкетонурии служит уровень фенилаланина в крови, который должен:
- в раннем возрасте составлять 120–240 мкмоль/л;
- у детей дошкольного возраста – не превышать 360 мкмоль/л;
- у школьников – не превышать 480 мкмоль/л;
- у детей старшего школьного возраста допустимо увеличение содержания фенилаланина в крови до 600 мкмоль/л.
Пищевой рацион строится путем резкого ограничения поступления белковых продуктов животного и растительного происхождения и, следовательно, фенилаланина. Для облегчения расчетов принято считать, что 1 г условного белка содержит 50 мг фенилаланина.
При лечении фенилкетонурии полностью исключают продукты, богатые белком и фенилаланином: мясо, рыбу, сыр, творог, яйца, бобовые и др. В пищевой рацион больных входят овощи, фрукты, соки, а также специальные малобелковые продукты – амилофены.
Для коррекции белкового питания и восполнения недостатка аминокислот при фенилкетонурии назначаются специальные лечебные продукты:
- белковые гидролизаты: нофелан (Польша), апонти (США), лофенолак (США);
- смеси L-аминокислот, лишенные фенилаланина, но содержащие все другие незаменимые аминокислоты: фенил-фри (США), тетрафен (Россия), П-АМ универсальный (Великобритания).
Несмотря на обогащение аминокислотных смесей и белковых гидролизатов минеральными и другими веществами, больные фенилкетонурией нуждаются в дополнительном назначении витаминов, в частности группы В, минеральных соединений, особенно содержащих кальций и фосфор, препаратов железа и микроэлементов.
В последние годы для страдающих фенилкетонурией была обоснована необходимость применения препаратов карнитина (L-карнитин, элькар в средней суточной дозе 10–20 мг/кг массы в течение 1–2 мес. 3–4 курса в год) для профилактики его недостаточности.
Параллельно лечение фенилкетонурии осуществляется медикаментозным патогенетическим и симптоматическим лечением ноотропными средствами, препаратами, улучшающими сосудистую микроциркуляцию, по показаниям – антиконвульсантами.
Широко используется лечебная гимнастика, общий массаж и др. Комплексная реабилитация детей с фенилкетонурией предусматривает специальные методы педагогических воздействий в процессе подготовки к школе и школьного обучения. Больные нуждаются в помощи логопеда, педагога, в ряде случаев – дефектолога.
Большие споры вызывает вопрос о длительности диетотерапии в лечении фенилкетонурии. В последнее время большинство врачей принимает точку зрения о необходимости продолжительного выполнения диетических рекомендаций. Обследование детей, прекративших соблюдать диету в школьном возрасте, и детей, продолжавших получать диетотерапию, однозначно показало значительно более высокий уровень интеллектуального развития последних.
У больных фенилкетонурией старшего возраста, в том числе подростков, безусловно, возможно постепенное расширение диеты в связи с улучшением толерантности к фенилаланину. Коррекция питания осуществляется, как правило, путем введения в рацион ограниченного количества круп, молока и некоторых других натуральных продуктов, содержащих относительно умеренное количество фенилаланина. В период расширения рациона проводятся оценка нервно-психического статуса детей, контроль электроэнцефалограммы, уровня фенилаланина в крови.
В возрасте старше 18–20 лет проводится дальнейшее расширение диеты, однако и во взрослом периоде пациентам рекомендуется отказаться от высокобелковых продуктов животного происхождения.
Особенно строго подходят к диетотерапии девочек, страдающих фенилкетонурией, и женщин в репродуктивном периоде. Такого рода больным фенилкетонурией необходимо продолжать диетическое лечение для обеспечения рождения здорового потомства.
В последние годы разрабатывается способ снижения уровня фенилаланина в крови путем приема препарата, содержащего фенилаланингидроксилазу растительного происхождения.
Данная статья опирается на статью из книги «Врожденные и наследственные заболевания» под редакцией профессора П.В.Новикова, М., 2007
Симптомы
У детей с фенилкетонурией светлые волосы, бледная кожа и голубые глаза.
Ребенок с фенилкетонурией рождается внешне здоровым, то есть ничем не отличается от других детей. С поступлением пищи в организм начинается попадание белка, а значит и фенилаланина. Последний постепенно накапливается, и обычно к 2 месяцам жизни появляются первые симптомы: вялость или беспокойство, отсутствие интереса к окружающему миру, срыгивания, изменения мышечного тонуса. Иногда срыгивания столь частые и обильные, что возникает подозрение на патологию желудочно-кишечного тракта (пилоростеноз). Ребенок из-за срыгиваний может плохо набирать в весе.
К 4-6 месяцам становится очевидной задержка психического развития. Ребенок не следит за игрушкой, не реагирует на звук, не узнает родителей. Чем дольше продолжается поступление фенилаланина в организм с едой, тем выраженнее нарушения в психической и мыслительной сферах. Развитие речи резко задерживается. Иногда словарный запас может ограничиваться несколькими словами. Если диагноз не будет выставлен и не будет начато лечение, то к 3-4 годам умственные нарушения достигнут степени идиотии (самая тяжелая степень олигофрении).
Особенностью клинического течения фенилкетонурии является необратимость возникших психических и интеллектуальных изменений. То есть при позднем выявлении помочь таким деткам уже нельзя – на всю жизнь они остаются умственно отсталыми.
Физическое развитие также отстает: дети позже начинают держать голову, переворачиваться, сидеть. Когда такие дети начинают ходить, то при этом они широко расставляют ножки, сгибая их одновременно в коленных и тазобедренных суставах. Походка покачивающаяся, мелкими шажками. В положении сидя дети принимают «позу портного» — сгибают и руки, и ноги, поджимая последние под себя. Обычно объем головы меньше, чем в норме. Может быть выраженная микроцефалия: маленькая голова.
Из других неврологических симптомов возможны нарушения мышечного тонуса, судорожные припадки. Эпилептические приступы обычно появляются в возрасте 1,5 лет и приводят к еще большему прогрессированию нарушений интеллекта.
У части больных фенилкетонурией появляются непроизвольные движения в конечностях, дрожание (гиперкинезы). В движениях нет плавности и согласованности, нарушается равновесие.
Кроме ряда психических и интеллектуальных изменений, фенилкетонурию характеризуют следующие симптомы:
- специфический «мышиный» запах (или запах плесени) от ребенка: этот симптом характерен только для фенилкетонурии. Запах появляется в результате выделения продуктов метаболизма фенилаланина (фенилпировиноградной, фенилмолочной, фенилуксусной кислот) через кожу и с мочой;
- кожные проявления: дерматиты, экзема, просто шелушение (возникают по той же причине, что и «мышиный» запах);
- позднее прорезывание зубов: у таких детей первые зубы могут появиться после 18 месяцев, эмаль недоразвита;
- нарушение пигментации: у таких детей обычно голубые глаза, очень светлая кожа и волосы в результате снижения количества меланина (его содержание зависит от метаболизма фенилаланина). Из-за этого у таких детей наблюдается повышенная чувствительность к солнечному свету;
- вегетативные симптомы: пониженное артериальное давление, повышенная потливость, запоры, акроцианоз (синюшность кистей и стоп);
- нередко фенилкетонурия сопровождается врожденными пороками сердца.
Атипичные случаи фенилкетонурии, связанные с нарушением деятельности других ферментов, участвующих в метаболизме фенилаланина, кроме умственных изменений характеризуются развитием мышечной слабости во всех конечностях с одновременным повышением мышечного тонуса, спастическим тетрапарезом. Также при этих формах развивается слюнотечение, приступы повышения температуры.
У взрослых людей, страдающих фенилкетонурией, возможно появление судорожных припадков, нарушений координации, дрожания в конечностях, ухудшения памяти и внимания, возникновение депрессии. Обычно подобные симптомы возникают при несоблюдении элиминационной диеты.
Общее описание
Фенилкетонурия (ФКУ) — это тяжкое наследуемое заболевание, происходящее вследствие неправильного обмена ароматических аминокислот и манифестирующееся олигофренией, задержкой физического развития, локомоторными расстройствами. ФКУ заболевает примерно один из 5-10 тыс. новорожденных. Среди больных преобладают девочки, т.к. младенцы мужского пола практически все погибают во время первого года жизни.
В результате того что при ФКУ не расщепляется аминокислота фенилаланин, поступающая с белковой пищей, в организме накапливаются соединения, обладающие нейротоксическим действием, следствием чего является развитие олигофрении. ФКУ наследственно детерминирована и возникает только при условии, что каждый из родителей передал ребенку патологический ген. Практически такое возможно при близкородственных браках. Считается, что порядка 2% популяции являются носителями такого типа гена, но при этом не имеют признаков заболевания.
Лечение
Болезнь фенилкетонурия в России лечится посредством диетотерапии, ограничивающей употребление белковых продуктов. В мире ведутся разработки препаратов, которые позволят контролировать уровень фенилаланина без соблюдения жесткой диеты. Перспективными направлениями исследований считаются:
- применение растительного фермента фенилаланинлиазы;
- генотерапия, направленная на коррекцию мутировавшего гена, вызывающего нарушения аминокислотного обмена;
- попытки введения гена фенилаланингидроксилазы в пораженные клетки печени.
Постоянное наблюдение у врачей (педиатра, невролога), соблюдение диеты, ограничивающей употребление белка, от рождения до половой зрелости является главным условием нормального развития ребенка, больного классической формой фенилкетонурии. Контроль уровня фенилаланина проводят регулярно: в первые 3 месяца жизни – один раз за неделю, от 3-х месяцев до года – минимум раз за месяц, от года до 3-х лет – 1 раз за два месяца, затем – по рекомендациям врача (около 4 раз за год). Количество употребляемых больным белков корректируется в зависимости от возраста и нагрузок.
Диетотерапия
Диета при фенилкетонурии основана на исключении из рациона больного животных белков. При ее постоянном строгом соблюдении удается предотвратить развитие церебральных нарушений, нарушений функции печени, если заболевание диагностировано в первые недели жизни. К строго запрещенным продуктам относятся:
- все виды мясных продуктов, колбасных изделий;
- все виды рыбы, морепродуктов;
- сыр;
- творог;
- яйца;
- орехи;
- хлебобулочные, кондитерские изделия;
- соевые продукты;
- крупы, хлопья.
Ежедневный рацион больного должен содержать овощи, фрукты, ягоды и зелень; разрешено употребление меда и сахара, крахмала, рисовой и кукурузной муки, сливочного, растительного масла. Ряд продуктов необходимо частично ограничить. С разрешения лечащего врача возможно употребление небольшого количества:
- молочных продуктов;
- картофеля, белокочанной капусты;
- овощных консервов.
Необходимые для развития и роста ребенка аминокислоты содержат специальные лечебные смеси, очищенные от лактозы, содержащие пептиды (молочные белки, уже расщепленные ферментами) и свободные аминокислоты (триптофан, тирозин, таурин, цистин, гистидин). Они производятся в форме порошков, которые разводят водой или сцеженным грудным молоком (при условии соблюдения матерью специальной диеты). Такими смесями и специальными продуктами для детей разного возраста являются:
- Афенилак;
- Мдмил-ФКУ-0;
- Аналог-СП;
- Апонти;
- Берлафен;
- Минафен;
- Лофенилак;
- Нофелан;
- Тетрафен;
- Циморган.
Медикаментозная терапия
Даже при регулярном получении белковых гидролизатов и аминокислотных смесей, больные фенилкетонурией нуждаются в дополнительном назначении комплексов витаминов (например, группы В) и минеральных соединений (содержащих препараты железа, фосфор и другие микроэлементы). Проводят симптоматическое и патогенетическое медикаментозное лечение с применением:
- препаратов карнитина для профилактики его дефицита (Элькар, L-карнитин);
- антиконвульсанты при судорожных припадках (Депакин, Клоназепам);
- ноотропные препараты для поддержания высших психических функций головного мозга (Пирацетам);
- средства, стимулирующие сосудистую микроциркуляцию;
- при атипичных формах фенилкетонурии, не поддающихся диетотерапии – гепатопротекторы, препараты с Леводопой, 5-окситриптофан, Тетрагидробиоптерин.
Широко применяется лечебная гимнастика, общий массаж. Реабилитация детей с фенилкетонурией предусматривает специальные методы педагогической подготовки во время дошкольного и школьного обучения. Больные нуждаются в регулярной помощи логопеда, педагога, дефектолога. Пациентам свойственна повышенная утомляемость, иногда требуется коррекция поведения, поэтому такие дети требуют к себе повышенного внимания и особого подхода.
Профилактика
Специфических методов профилактики ФКУ не существует. Но некоторые шаги, все же, можно сделать для оценки рисков, и принятия необходимых мер. Они заключаются в:
- Генетическом консультировании. Оно необходимо людям, которые сами страдают от ФКУ, или имеют к ней наследственную предрасположенность, и планируют в ближайшем времени завести ребенка. Также консультация важна и в случае, когда в семье уже родился больной малыш. Проводит ее врач-генетик, который должен разобраться, как происходила передача гена, ответственного за ФКУ, в прошлых поколениях.
- Скрининге новорожденных. Такое исследование не поможет предотвратить ФКУ, но позволит выявить ее как можно раньше – прежде, чем болезнь начнет провоцировать гибель клеток головного мозга.
- Регулярном прохождении врачебной консультации и диете. Прежде всего, это касается женщин, страдающих от ФКУ. Все нюансы, касающиеся планирования или протекания беременности, соблюдения диеты до, во время и после этого периода необходимо обязательно уточнять у лечащего врача.