Причины, симптомы и лечение болезни вильсона-коновалова
Содержание:
Данные статистики
Последнее время диагностируют больше случаев заболевания. 0,56% населения Земли являются носителями патологически измененного гена.
В тех регионах, где практикуют браки между близкими родственниками, частота заболевания выше. Заболеваемость не зависит от половой принадлежности. Но у детей и молодежи оно диагностируется чаще. Симптомы могут проявиться к 19-20 годам. До пяти лет симптомы могут полностью отсутствовать.
Если патологию не лечить, летальность составляет 100%. Такие больные редко доживают до 30-ти. Причина летальности – геморрагические осложнения, печеночная, почечная недостаточность.
Патогенез
Медь выполняет множество функций в организме. В основном она выступает в качестве кофактора для некоторых ферментов, таких как церулоплазмин, цитохром с-оксидаза, дофамин бета гидроксилаза, супероксиддисмутаза и тирозиназа.
Медь всасывается из желудочно-кишечного тракта. Транспортный белок на клетках тонкой кишки CMT1 (англ. Copper Membrane Transporter 1) перемещает медь внутрь клеток. Часть меди связывается с металлотионеином, а другая — перемещается в сеть Гольджи с помощью транспортного белка ATOX1. В аппарате Гольджи в ответ на повышение концентрации меди фермент ATP7A (англ. Copper-transporting ATPase 1) высвобождает этот элемент через воротную вену в печень. В печёночных клетках белок ATP7B связывает медь с церулоплазмином и высвобождает его в кровь, а также удаляет избыток меди с выделяющейся жёлчью. Обе функции ATP7B нарушены при болезни Вильсона. Медь накапливается в ткани печени; церулоплазмин продолжает выделяться, но с недостатком меди (апоцерулоплазмин) и быстро разрушается в кровотоке.
Когда меди в печени становится больше, чем белков её связывающих, происходит их окислительное повреждение за счёт реакции Фентона. Это приводит к воспалению печени, её фиброзу и в итоге к циррозу. Также из печени в кровоток выделяется медь, которая не связана с церулоплазмином. Эта свободная медь оседает по всему организму, особенно в почках, глазах и головном мозге.
Основную роль в патогенезе играет нарушение обмена меди, её накопление в нервной (особенно поражены базальные ганглии), почечной, печёночной ткани и роговице, а также токсическое повреждение медью данных органов. Нарушение метаболизма выражается в нарушении синтеза и снижении в крови концентрации церулоплазмина. Церулоплазмин участвует в процессе выведения меди из организма. В печени формируется крупноузловой или смешанный цирроз. В почках в первую очередь страдают проксимальные канальцы. В головном мозге поражаются в большей степени базальные ганглии, зубчатое ядро мозжечка и черная субстанция. Отложение меди в десцеметовой мембране глаза приводит к формированию кольца Кайзера-Флейшера.
Лечение
Схема лечения Вильсона-Коновалова подбирается в индивидуальном порядке. Основная задача медикаментозной терапии заключается в выведении избыточного количества меди из организма пациента.
Программа лечения от болезни Вильсона-Коновалова состоит из следующих этапов:
- Диагностика со сбором требуемых анализов и тестов;
- Получение консультации у специалистов с назначением индивидуальной схемы лечения;
- Осуществление медикаментозных и лечебных процедур для устранения симптомов заболевания;
- Прохождение реабилитационных мер в отделении неврологии.
Основные методы лечения Вильсона-Коновалова:
- Прием Д-пенициламина, уменьшающего количество меди в кровяных тельцах больного. Данный вариант лечения относится к патогенетическому;
- Реабилитационные меры в отделении неврологии для устранения негативного воздействия на нервную систему больного;
- Внутренний прием сорбентов и гепатопротекторов (при наличии повреждений печеночных тканей);
- Трансплантация печени при наличии серьезных, не возобновляемых повреждений органа;
- С помощью специализированного оборудования осуществляются процедуры по очистке плазмы крови от токсинов и свободной меди (гемоиммуносорбция), удалению из плазмы продуктов патологического распада (мембранный плазмаферез). Пациенту могут быть назначены до 3 процедур п очистке крови. Данная методика лечения зарекомендовала себя, как максимально эффективная.
Первые результаты от очистки крови заметны уже после 14 календарных дней. Медикаментозное лечение может длиться до полугода.
Кроме того, любая схема лечения от болезни Вильсона-Коновалова сопровождается приемом медикаментов. Среди них чаще всего выписывают:
Лекарственные препараты, снижающие количество меди в организме больного (хелаты);
Медикаменты, способствующие регенерации печеночных клеток;
Желчегонные лекарственные медикаменты;
Комплекс витаминов (важно применение В6);
Лекарства, блокирующие поступление меди в организм с продуктами питания (например, содержащие цинк);
Медикаменты с антиоксидантными свойствами, снижающие интоксикацию организма;
Средства, подавляющие действие иммунной системы;
Лекарственные препараты успокоительного характера для устранения неврологических расстройств;
Средства, обладающие противовоспалительным результатом.
Назначенные доктором медикаменты пациент обязан принимать с начала диагностирования болезни и до конца его жизни. Если больной снизит дозировку или полностью прекратит лечение, то Вильсон-Коновалов может вернуться с более сильной симптоматикой.
В запущенных случаях заболевания, когда медикаментозные методы не дают должного результата, а состояние больного ухудшается диагностирована печеночная недостаточность, назначается трансплантация печени. Новая печень приживается и нормально функционирует уже через месяц после операции.
Терапия патологии
Болезнь Вильсона сопровождается увеличением количества меди в кровотоке. Лечение разрабатывается таким образом, чтобы снизить ее концентрацию. Пациентам показано придерживаться диетического питания, которое заключается в исключении богатых медью продуктов. Пациентам не рекомендуется употребление изделий из какао и шоколада. Они должны отказаться от грибов и печени. Употребление любых видов орехов специалисты запрещают.
В период протекания болезни рекомендовано применение медикаментозной терапии. Наиболее часто больным делают назначение Пеницилламина. Суточная дозировка препарата составляет 1,2-2 грамма. Она определяется доктором в соответствии с индивидуальными особенностями пациента и степенью тяжести протекания патологии. При регулярном применении лекарства можно добиться стойкого клинического улучшения и даже полного устранения симптоматики. Параллельно рекомендовано принимать препараты, в состав которых входит большое количество витамина В6.
Лечение Пеницилламином требует придерживаться специальной схемы. На начальных этапах больному назначают 150 миллиграмм препарата через день. Такой схемы пациент должен придерживаться 7 дней. Следующую неделю лекарство принимается в такой же дозировке ежедневно. Далее рекомендовано каждую неделю повышать суточную дозировку медикамента на 150 миллиграмм. Максимальная доза препарата составляет 2 грамма. После улучшения состояния больного рекомендуется придерживаться поддерживающей дозировки 450-600 миллиграмм в сутки. Витамин В6 принимается в дозе 25-50 миллиграмм.
При возникновении нежелательных эффектов в виде тошноты, аллергии, болезней почек не рекомендован прием Пеницилламина. После этого прием препарата проводится в минимальной дозировке. Одновременно рекомендуется принимать 20 миллиграмм Преднизолона ежедневно. Длительность терапии заболевания составляет 10 суток. Если организм человека не принимает Пеницилламин, то рекомендовано назначение цинка Сульфата. Его необходимо принимать три раза в день по 200 миллиграмм.
Если у пациента диагностируется непереносимость Пеницилламина, то ему показан прием Унитола. Прием лекарство должен проводиться в течение месяца. После этого делают перерыв на 3 месяца. При использовании лекарства состояние человека улучшается, а симптоматика болезни стихает. Если в период протекания патологического процесса доминируют гиперкинезы, то больному рекомендовано принимать нейролептики короткими курсами. При возникновении ригидности пациентам показана:
- Леводопа;
- Тригексифенидил;
- Карбидопа.
Если консервативное лечение патологии не приносит желаемых результатов, а заболевание у пациентов протекает в тяжелой форме, то рекомендовано проведение хирургического вмешательства. В современных зарубежных клиниках проводятся операции по пересадке печени. Если исход хирургии положителен, то это приводит к улучшению состояния пациента и восстановлению процесса обмена меди в организме. В дальнейшем для терапии патологического процесса рекомендовано применение иммуносупрессивной терапии. В клинической практике нашей страны все чаще применяется биогенмоперфузия. При этом методе используются изолированные живые клетки печени и селезенки. При условии правильного проведения процедуры будет гарантирована ее высокая эффективность.
Схема лечения при патологии должна назначаться только доктором, что положительно отобразится на результате.
Клиническая картина и течение
Гепато-церебральная дистрофия начинается в детском или молодом возрасте и имеет хроническое прогрессирующее течение. Во многих случаях появлению симптомов поражения нервной системы предшествуют висцеральные расстройства в виде нарушения деятельности печени и желудочно-кишечных расстройств (желтуха, боли в правом подреберье, диспептические явления). Порой развивается выраженный гепато-лиенальный синдром. Со стороны нервной системы на первый план выступают экстрапирамидные симптомы в виде мышечной ригидности, гиперкинезов и расстройств психики. Пирамидные симптомы могут быть, но чаще отсутствуют. Чувствительность обычно не нарушена.
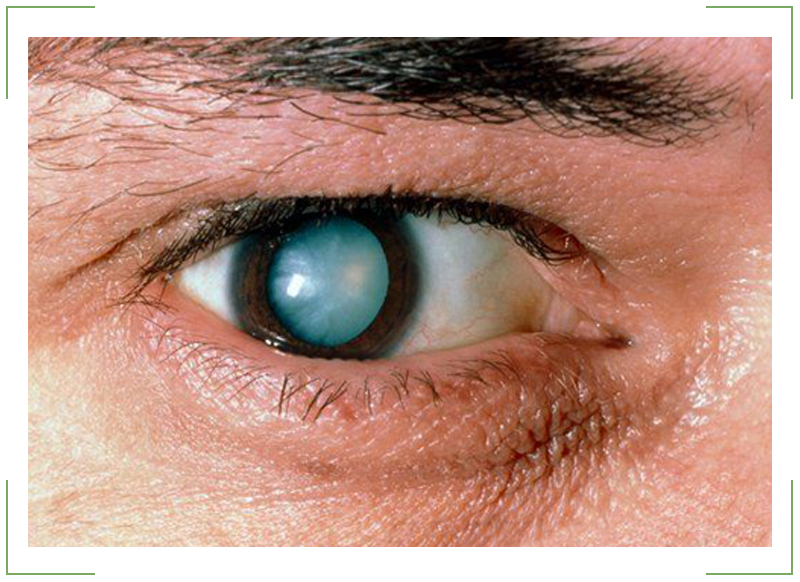
Типичным симптомом болезни являются кольца Кайзера-Флейшера — отложения по периферии роговой оболочки содержащего медь зеленовато-бурого пигмента, более выраженные на поздних стадиях. Иногда отмечается желтовато-коричневая пигментация кожи туловища и лица. Часты геморрагические явления (кровоточивость дёсен, носовые кровотечения, положительная проба жгута), мраморность кожи, акроцианоз. Капилляроскопия обнаруживает атонию капилляров и застойность кровотока. Отмечаются суставные боли, профузные поты, остеопороз, ломкость костей.
Патология печени клинически выявляется примерно у 30 % больных, а в ряде случаев она может быть обнаружена только функциональными пробами, например пробой с нагрузкой галактозой, пробой Квинка, пробой Бергмана-Эльботта, бромсульфофталеиновой пробой; количество билирубина в крови и уробилина в моче обычно увеличено; изменены осадочные реакции Таката-Ара и Грея, обычны лейкопения, тромбоцитопения, гипохромная анемия.
Различают 5 форм гепато-церебральной дистрофии:[уточнить]
- Брюшная форма — тяжёлое поражение печени, приводящее к смерти раньше появления симптомов со стороны нервной системы; заболевают дети. Её продолжительность от нескольких месяцев до 3-5 лет.
- Ригидно-аритмогиперкинетическая, или ранняя форма — отличается быстрым течением; начинается также в детском возрасте. В клинической картине преобладают мышечная ригидность, приводящая к контрактурам, бедность и замедленность движений, хореоатетоидные или торсионные насильственные движения. Характерны дизартрия и дисфагия, судорожный смех и плач, аффективные расстройства и умеренное снижение интеллекта. Заболевание длится 2-3 года, заканчивается летально.
- Дрожательно-ригидная форма встречается чаще других; начинается в юношеском возрасте, течёт медленнее, порой с ремиссиями и внезапными ухудшениями, сопровождающимися субфебрильной температурой; характеризуется одновременным развитием тяжёлой ригидности и дрожания, дрожание очень ритмичное (2-8 дрожаний в секунду), резко усиливается при статическом напряжении мышц, движениях и волнении, в покое и во сне исчезает. Иногда обнаруживаются атетоидные хореоформные насильственные движения; наблюдаются также дисфагия и дизартрия. Средняя продолжительность жизни около шести лет.
- Дрожательная форма начинается в возрасте 20-30 лет, течёт довольно медленно(10-15 лет и больше); дрожание резко преобладает, ригидность появляется лишь в конце болезни, а порой наблюдается гипотония мышц; отмечается амимия, медленная монотонная речь, тяжёлые изменения психики, часты аффективные вспышки. Наблюдаются эпилептиформные припадки.
- Экстрапирамидно-корковая форма встречается реже других форм. Типичные для гепато-церебральной дистрофии нарушения в дальнейшем осложняются апоплектиформно развивающимися пирамидными парезами, эпилептиформными припадками и тяжёлым слабоумием (обнаруживаются обширные размягчения в коре больших полушарий). Длится 6-8 лет, заканчивается летально.
Наибольшая летальность (50 %) отмечается при печёночной форме с массивным некрозом и гемолизом у детей до 6 лет. Смерть больных от неврологических нарушений при отсутствии лечения наступает через 5-14 лет. Основная причина при этом интеркуррентные заболевания или желудочно-кишечные кровотечения, портальная гипертензия.
Причины болезни Вильсона
Болезнь Вильсона — наследственное, поддающееся лечению заболевание, при котором происходит постепенное накопление меди в печени, головном мозге (преимущественно в базальных ядрах), роговице, почках, что вызывает тяжелые функциональные нарушения, ведущие к необратимым повреждениям. Без лечения заболевание заканчивается смертью, однако своевременная постановка диагноза и лечение устраняют или предотвращают его симптомы.
Болезнь Вильсона наследуется аутосомно-рецессивно. Распространенность гетерозигот по мутантному гену составляет 1:200, а гомозигот — 1:30 000. Ген, ответственный за болезнь Вильсона, расположен на 13-й хромосоме вблизи гена, кодирующего эстеразу D. У 95% больных наблюдается дефицит или полное отсутствие церулоплазмина (сывороточный белок, играющий главную роль в транспорте меди). Это обусловлено снижением транскрипции гена церулоплазмина, расположенного на 3-й хромосоме.
Медь (Cu) — важнейший микроэлемент, который входит в состав ферментов, например цитохромоксидазы, тирозиназы, супероксиддисмутазы и др.
Метаболизм меди. Поступление Cu в организм в норме составляет приблизительно 2-5 мг в сутки, из которых 40-60 % всасываются в желудке и верхнем отделе двенадцатиперстной кишки. С помощью переносчика Си поступает в клетки печени, связывается с белками или включается в состав церулоплазмина (ферроксидаза), который относительно прочно связывает шесть атомов Cu. В комплексе с церулоплазмином Cu выделяется в плазму (приблизительно 93 % от содержания в плазме), где она окисляет Fe2+ в Fe3+. Незначительное количество Cu, связанное с церулоплазмином, высвобождается в периферических тканях. Экскреция Cu в желчь осуществляется с помощью АТФазы Р-типа, которая называется Cu-АТФаза (АТР7В). «Состарившийся», не содержащий сиаловых кислот церулоплазмин разрушается в печени, Си высвобождается, прочно связывается с белками желчи и выводится из организма с калом. В сутки выделяется приблизительно 1,2 мг меди.
Болезнь Вильсона (гепатолентикулярная дегенерация) — аутосомно-рецессивное нарушение метаболизма Cu, при котором в печени, ЦНС, глазах и других органах происходит ее избыточное отложение. Заболевание вызывается мутациями гена АТР7В, кодирующего белок CU-ATP7B. Мутация приводит к снижению выведения значительного количества Cu с желчью и уменьшению включения Cu в церулоплазмин. В результате при общей концентрации Си ниже нормы в печени и затем в плазме накапливается свободная или слабо связанная медь. В такой форме Cu токсична, поскольку связывается преимущественно с сульфгидрильными группами белков и способствует образованию свободных радикалов O2 и перекисному окислению липидов.
Накопление свободной Cu вызывает развитие анемии и хронического гепатита, который впоследствии приводит к циррозу. При молниеносном гепатите из некротически измененной ткани печени внезапно высвобождается большое количество Cu, что может вызвать гемолитический криз. Накопление Cu в ЦНС приводит к многочисленным и разнообразным неврологическим, нервно-мышечным и психогенным расстройствам. Отложение Cu в виде зернистой массы в десцеметовой оболочке глаз способствует образованию кольца Кайзера—Флейшера по периферии роговицы. В патологический процесс могут вовлекаться почки, скелет и сердце. Поскольку избыточное отложение Си обусловлено мутацией гена АТР7В, излечение от болезни Вильсона возможно путем трансплантации печени.
Лечение синдрома Вильсона-Коновалова
Для лечения синдрома Вильсона-Коновалова сейчас активно используется D—пеницилламин, известный и под названием купренил. Эффективность лечения возможна лишь при терапии, направленной на снижение меди в организме, начатой на ранней стадии развития синдрома Вильсона-Коновалова, при запущенных формах данное заболевание полноценному лечению не поддается.
Для профилактики ее дальнейшего развития необходим регулярный мониторинг, позволяющий правильно определить содержание в организме цинка, меди и уровня церулоплазмина. Необходима регулярная диагностика синдрома Вильсона-Коновалова, обследование у врача должно сопровождать своевременным забором крови на биохимический и общий анализ, больной нуждается в постоянном контроле со стороны терапевта и невропатолога.
Большинству пациентов, страдающих синдромом, регулярное лечение синдрома Вильсона-Коновалова проводят препаратом D–пеницилламином, он помогает чувствовать себя практически нормально, вести полноценный образ жизни, даже работать. Но от потребления данного препарата у некоторых пациентов сопровождается такими побочными эффектами, как волчаночноподобный синдром (системная красная волчанка), проявления тошноты, нефротический синдром, пемфигус.
Доктора
специализация: Гепатолог / Невролог / Гастроэнтеролог
Сафонов Игорь Дмитриевич
6 отзывовЗаписаться
Подобрать врача и записаться на прием
Купренил
Унитиол
Цинктерал
Мирапекс
Гепа-Мерц
Верошпирон
Тиоктацид
Глиатилин
Возможные осложнения
В связи с тем, что болезнь поражает печень, нервную систему, вероятные осложнения делятся на такие основные группы:
- Тяжелые заболевания печени. К одним из них относят цирроз, возникающий у большинства пациентов. Прогрессирует он медленно, сопровождается желтушностью кожных покровов, деформацией пальцев рук и ног, расширенными венами на передней брюшной стенке, отеками голеней. Часто больные страдают от кровотечений, возникающих в желудке. Развивается печеночная недостаточность, симптомами которой являются сонливость, поведенческие расстройства, на последней стадии — кома.
- Смерть. Летальный исход ожидает более 70% больных, страдающих от печеночной недостаточности, в особенности — фульминантной.
- Неврологические нарушения. Сюда относят мышечную дистонию, дизартрию, расстройства личности и поведения, эпилептические припадки.
- Невозможность забеременеть у женщин.

Диагностика болезни Вильсона — Коновалова
Диагноз основывается на сочетании клинических симптомов, лабораторных данных и молекулярно-генетического тестирования. Ни один лабораторный тест, за исключением определения болезнетворного гена АТР7В на молекулярном уровне, не обеспечивает 100 % гарантию диагностики заболевания.
Основные диагностические показатели болезни Вильсона — Коновалова:
- Церулоплазмин: снижение на 50 %. Может быть нормальным. По другим данным — менее 20 мг/дл. По ряду причин (болезнь Менкеса, печёночная недостаточность, нефротический синдром, длительное парентеральное питание и др.) анализ может быть ложноотрицальным.
- Суточная экскреция меди с мочой: >100 мкг/сут., или > 40 мкг/сут. у детей. При бессимптомном течении показатели не превышают норму 40 мкг/сут.
- «Свободная» медь сыворотки: > 1,6 мкМ/л
- Медь в ткани печени: > 4 мкМ/г или > 250 мкг/г сухого веса.
| Симптомы | Лабораторные тесты |
|---|---|
| 1. Кольца Кайзера-Флейшера:• присутствуют (0 баллов)• отсутствуют (2 балла) | 1. Экскреция (выделение) меди с мочой:• норма (0 баллов)• 1-2 нормы (1 балл)• более 2 норм (2 балла)• норма, но увеличение более 5 норм при пробе с купренилом (2 балла) |
| 2.Нейропсихиатрическая симптоматика: (изменения на МРТ)• присутствует (2 балла) • отсутствует (0 баллов) | 2. Количественное определение меди в биоптатах печени:• норма (-1 балл) • 50-250 мкг/г (1 балл)• Более 250 мкг/г (2 балла) |
| 3. Кумбс-негативная гемолитическая анемия:• присутствует (1 балл) • отсутствует (0 баллов) | 3. Родамин – позитивные гепатоциты (при невозможности количественного определения меди в печени):• присутствуют (0 баллов) • отсутствуют (1 балл) |
| 4. Уровень церулоплазмина в сыворотке крови (при норме более 20 мг/дл):• норма (0 баллов) • 10-20 (1 балл) • менее 10 (2 балла) | |
| Молекулярно-генетическое исследование (выявление мутаций гена АТР7В) • гомозигота (два одинаковых гена определяющих в данном случае проявление болезни) или компаунд-гетерозигота, (из пары генов оба имеют разные мутации, приводящие к болезни, но они не идентичны друг другу) (4 балла)• гетерозигота (один ген из пары является нормальным, один с мутацией) (1 балл)• мутаций не обнаружено (0 баллов) |
Суммарные баллы:
- 4 и более — высокая вероятность болезни;
- 2-3 — болезнь вероятна, но требуется дальнейшее обследование пациента;
- 0-1 — болезнь сомнительна.
Для уточнения степени поражения и формы заболевания используются МРТ головного мозга, хотя только на основании МРТ диагноз поставить нельзя. При МРТ исследовании сразу видны характерные очаги и уменьшение объёма головного мозга. Специфичным, но более редко встречающимся симптомом при данном заболевании на МРТ снимке является картина, напоминающая «лицо гигантской панды». КТ- и МРТ-проявления могут опережать клиническую симптоматику.
Компьютерная томография (КТ) головного мозга при наличии болезни выявляет увеличение желудочков, атрофию коры и ствола мозга. Однако более важным диагностическим методом при церебральной форме заболевания является именно МРТ.
Для определения очагов скопления меди и нехирургической оценки метаболизма мозга может потребоваться магнитно-резонансная спектроскопия (МРС). Это метод, позволяющий оценить изменения биохимической концентрации веществ при различных заболеваниях в тканях организма.
Также используется позитронно-эмиссионная томография (ПЭТ), позволяющая определить степень обмена и транспорта веществ в организме.
Перспективным методом ранней диагностики является транскраниальное УЗИ головного мозга.
Изменения, определяемые при УЗИ, КТ и МРТ печени и почек определяются и при других болезненных состояниях, поэтому не являются строго специфичными для болезни Вильсона — Коновалова и могут быть использованы только для оценки эффективности лечения.
Новым методом диагностики степени выраженности цирроза (фиброза) печени является эластометрия печени. Исследование использует способность ультразвука проходить с различной скоростью через ткани разной плотности, что позволяет определить изменение нормальной плотности органа .
Диагностика болезни Вильсона
Сывороточная концентрация церулоплазмина при болезни Вильсона в 95% случаев ниже 1,3 мкмоль/л. Однако этого мало, чтобы поставить диагноз болезни Вильсона — примерно у 20% гетерозигот по мутантному гену количество церулоплазмина тоже снижено. При остром некрозе печени и у 15% больных с поражением печени как единственным проявлением заболевания концентрация церулоплазмина, являющегося белком острой фазы воспаления, может быть несколько повышена.
Концентрация меди в сыворотке. Так как церулоплазмин является основным белком, отвечающим за транспорт меди в крови, общая концентрация меди в сыворотке при болезни Вильсона часто снижена, однако концентрация свободной меди повышена, что и способствует ее отложению в различных тканях. Определение концентрации свобод ной меди в сыворотке — наиболее надежный метод предварительной диагностики болезни Вильсона. Ее вычисляют как разницу между общим содержанием меди в сыворотке и ее количеством, связанным с церулоплазмином.
Экскреция меди с мочой. Свободная медь в сыворотке легко выводится почками, поэтому при болезни Вильсона экскреция меди с мочой увеличена.
Биопсия печени. Для получения достоверного результата образец ткани должен быть достаточно большим (желательно, чтобы длина столбика ткани была не менее 1 см) и не загрязненным следами меди (использование одноразовых игл для биопсии снижает этот риск). Биопсия трансъюгулярным доступом не позволяет получить количество ткани, достаточное для количественного анализа. Другие заболевания, в частности первичный и вторичный билиарный цирроз, длительная обструкция желчного протока, также могут сильно повысить содержание меди в печени за счет нарушения ее экскреции с желчью. Однако у этих больных повышен уровень церулоплазмина в сыворотке.
В отдельных случаях при нормальном содержании церулоплазмина в сыворотке и невозможности провести биопсию из-за нарушений свертывания крови проводят нагрузочную пробу с изотопом меди 64Cu, имеющим период полураспада 12,8 ч. Препарат принимают внутрь (2 мг) или вводят в/в (500 мг), после чего отмечают зависимость концентрации радиоактивной меди в сыворотке от времени.
Если болезни Вильсона нет, радиоактивная медь появляется в сыворотке и снова исчезает через 4—6 ч. После того как изотоп захватывается печенью и связывается с вновь синтезированным церулоплазмином, наблюдается второй пик радиоактивности в сыворотке. При болезни Вильсона второй пик отсутствует, поскольку количество захватываемой для связывания с церулоплазмином меди снижено.
Кольца Кайзера—Флейшера всегда обнаруживают больных с неврологическими проявлениями болезни Вильсона; если имеется только поражение печени, они могут отсутствовать. Если кольца Кайзера—Флейшера не видны невооруженным глазом, проводят исследование с помощью щелевой лампы.
Для острого некроза печени при болезни Вильсона характерно сочетание низкой активности ЩФ и лишь небольшого повышения активности аминотрансфераз с желтухой, клиническими и гистологическими признаками некроза печени. Диагностическую ценность представляет также низкое отношение активности ЩФ к уровню общего билирубина.
Все братья и сестры больных должны пройти обследование на наличие у них болезни Вильсона. Проводится физикальное исследование.