Синдром трисомии х продолжительность жизни
Содержание:
- Клиническое описание
- Трисомия по Х хромосоме. Причины
- Цитологическая диагностика
- История исследования[править | править код]
- Лечение синдрома Патау
- Трисомия 21. Патофизиология
- Кольцевая 22 хромосома. Лечение
- Синдром Тернера, или моносомия Х-хромосомы
- Причины генетической патологии
- Трисомия по Х хромосоме. Причины
- Патофизиология
- Лечение
- Диагностика трисомии 21
Клиническое описание
Клиническая картина характеризуется недостаточностью пренатального роста, специфическими черепно-лицевыми особенностями, другими незначительными аномалиями, основными пороками развития, выраженной задержкой психомоторного и когнитивного развития.
Задержка роста начинается в пренатальном периоде, продолжается после рождения. Часто она связана со сложностями питания, которые могут потребовать энтерального вмешательства. Обычно присутствует микроцефалия послеродового периода.
Типичные краниофациальные функции включают долихоцефалию, короткие глазные щели, микрогнатию, внешние аномалии ушей, избыточную кожу на задней части шеи.
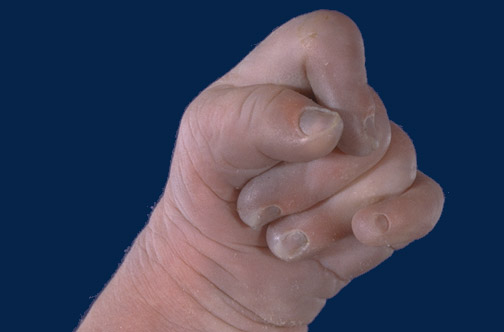
Другие характерные клинические признаки – сжатый кулак с переопределением пальцев (указательный палец перекрывает третий, 5 – й палец перекрывает 4). Особенно отличительны, маленькие ногти, недоразвитые пальцы руки, изогнутые ноги.
Наличие основных пороков развития распространено. Любой орган и система могут быть затронуты. Структурные дефекты сердца встречаются у более чем 90%.
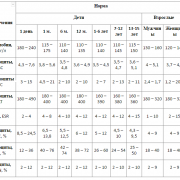
Таблица 1
Общие основные структурные пороки развития
| частота | Орган / Система | Преобладающий тип мальформации |
| Общий (> 75%) | сердце | дефекты перегородки, артериальный прототип протоков, поливаскулярная болезнь |
| Частые (25-75%) | Мочеполовая система | подковообразная почка |
| Менее часто (5-25%) | желудочно-кишечный тракт | omphalocele, атрезия пищевода с трахео-пищеводным фистулом, пилорический стеноз, Meckel diverticulum |
| Центральная нервная система | гипоплазия мозжечка, агенезис мозолистого тела, полимикрогирия, расщепление позвоночника | |
| черепно-лицевой отдел | орофациальные расщелины | |
| глаза | микрофтальмия, колобома, катаракта, непрозрачность роговицы | |
| конечности | радиальная аплазия, гипоплазия |
Трисомия по Х хромосоме. Причины
Трисомии по Х хромосоме представляют собой хромосомные аномалии, которые характеризуются наличием дополнительной Х-хромосомы. Несмотря на то, что трисомия X является генетическим нарушением, она не наследуется. Наличие дополнительной Х-хромосомы является следствием ошибки при расхождении хромосом. Эти ошибки возникают случайно и без видимой причины (спорадически). В большинстве случаев, дополнительная Х-хромосома имеет материнское происхождение. Примерно в 20 процентах случаев, нерасхождение происходит после зачатия. Исследователи считают, что симптомы и физические особенности этого расстройства связаны с сверхэкспрессией генов.
Цитологическая диагностика
Флюоресцентная гибридизация используется с целью выявления субмикроскопических делеций и дупликаций хромосом, которые являются слишком маленькими для идентификации методами конвенциональной цитогенетики. Ее также используют для идентификации «мягких» транслокаций и маркерных хромосом. РИ8Н-анализ выполняется на препарате метафазных хромосом, выделенных из культивируемых лимфоцитов, амниоцитов, ворсинок хориона, интерфазных ядер из крови, тканей, ворсинок хориона, амниотической жидкости при пренатальном выявлении аномалий развития плода и необходимости скрининга на анеуплоидии (трисомия 21 и т.д.). РИ8Н-анализ применяется с целью преимплантационной генетической диагностики по выявлению сбалансированных транслокаций или делеций. РИ8Н-метод предоставляет информацию по анализу специфической хромосомы или нескольких хромосом, но не выполняется для определения кариотипа.
История исследования[править | править код]
Синдром был предсказан в работах Патрисии Джейкобс 1959 года по XXY-синдрому, а впервые обнаружен в 1961 году при случайном обследовании мужчины, дети которого имели ряд заболеваний (в частности, один из детей имел синдром Дауна). XYY-синдром был описан как так называемый «синдром сверх-самца» или «синдром сверх-мужчины» (англ. super-male syndrome), при этом носителям синдрома приписывалось агрессивное поведение и тенденция к криминальным действиям. Первые исследователи болезни в 1960-х годах обнаружили относительно высокое количество мужчин с этим синдромом среди обитателей тюрем и психиатрических клиник. Это послужило основой для стереотипа о «сверх-мужчине».
Последующие исследования показали, что абсолютное большинство носителей синдрома никогда не имели отношения к преступности или психическим заболеваниям, но могут иметь повышенный риск проблем с обучением. Дополнительная Y-хромосома сама по себе не ведёт к чрезмерной агрессивности.
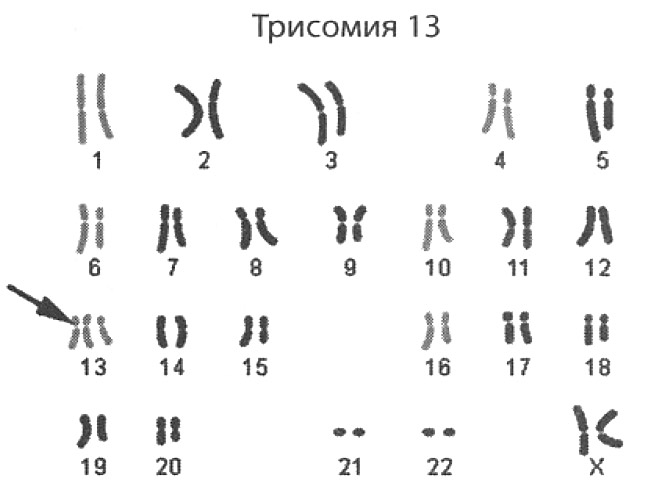
Лечение синдрома Патау
Выявить синдром Патау у плода можно на основании ультразвукового исследования. Патология определяется начиная с 20 недели беременности. Поводом для постановки предварительного диагноза выступают различные нарушения развития ребенка. При помощи исследования врач может заметить, что у плода присутствуют неправильный череп, заячья губа, низкая масса тела, отсутствуют волосы и имеет место быть симптомы, указывающие на развитие отклонений. Дополнительно могут проводиться следующие исследования:
- биопсия ворсин хориона;
- кордоцентез;
- амниоцентез;
- генетическое исследование наследственного материала.
На протяжении всей беременности женщина проходит 3 скрининга. Ей предстоит сдать венозную кровь для биохимического анализа. Первый скрининг выполняется на 11-14 неделе беременности. Аналогичную процедуру повторяют на 16-18 неделе. Самый последний скрининг проводится на 32-34 недели.
Изначально в крови определяют концентрацию ряда гормонов. Ими являются афп, ХГЧ и свободный эстриол. Показатели дают возможность выявить синдром Патау, Дауна и Эдвардса. Один лишь скрининг не позволяет со стопроцентной гарантией сказать о наличии заболевания у плода. Потому в обязательном порядке проводится УЗИ. На более поздних сроках производится забор амниотической жидкости. Если опасения подтверждаются, беременной женщине рекомендуется сделать аборт.
Обычно синдром Патау выявляется на УЗИ, который проводится после 20 недели беременности. Однако заметить патологию можно и во время исследований на 12 неделе. Несмотря на то, что плод еще маленький, нарушения уже можно заметить. УЗИ в этот промежуток времени позволяет определить:
- воротниковое пространство;
- длину костей;
- величину головы;
- симметричность полушарий мозга;
- присутствие основных органов;
- окружность живота.
Если у ребенка присутствует хромосомное нарушение, обусловленное синдромом Патау, у плода будет маленькая голова. Полушария мозга при этом несимметричные. Дополнительно может меняться размер носовой кости. Часто обнаруживаются лишние пальцы. Если на УЗИ заметно что-то странное, беременную направляют на иные исследования. Рекомендуется получить консультацию генетика.
Вылечить синдром Патау невозможно. Патология приводит к возникновению нарушений в развитии и строении внутренних органов. Медики могут попытаться облегчить состояние ребенка. Для этого проводится оперативное вмешательство. Чаще всего при синдроме Патау выполняется:
- Пластика лица. У многих пациентов при этом недуге наблюдаются расщелины на губах. Врачи устраняют этот недостаток.
- Удаление дополнительной матки. Аналогичное действие выполняется в отношении других подобных патологий, если они имеют место быть.
- Проводятся операции на внутренних органах. Обычно лечение осуществляется в отношении почек, мочеточников и сердца. Это позволяет облегчить уход за ребёнком.
Дополнительно проводится симптоматическое лечение. Врачи устраняют сопутствующие симптомы и укрепляют иммунитет ребенка. Действия необходимо выполнять для того, чтобы избежать воспаления органов. Дети с синдромом Патау всегда недоразвитые. Это касается физического и умственного развития. Полноценной жизнью жить такие пациенты вряд ли могут. Ребёнок не будет самостоятельным. Именно поэтому врачи рекомендуют прервать беременность на сроке до 22 недель.
Трисомия 21. Патофизиология
Дополнительная хромосома 21 затрагивает почти каждую систему органов, в результаты чего, у человека могут развиться проявления из широкого спектра фенотипических особенностей трисомии 21. Эти особенности включают в себя опасные для жизни осложнения, клинически значимые для жизни осложнения (например, тяжелая умственная отсталость) и изменения некоторых частей тела в размерах и в форме. Живорожденные почти всегда имеют задержки в физическом развитии, в развитии костей и зубов.
Сегодея существует две различных гипотезы, которыми можно объяснить механизм, по которому наличие лишних генов может привести к развитию таких аномалий. (1) Потеря хромосомного баланса и (2) эффект дозы гена. В соответствии с гипотезой эффекта дозы гена, гены, расположенные на хромосоме 21 могут сверхэкспрессироваться в клетках и тканях пациентов, что может внести свой вклад в развитие фенотипических аномалий.
Видимо, именно дополнительный экземпляр проксимальной части 21q22.3 приводит к развитию типичного фенотипа, который включает в себя:
- Умственная отсталость – большинство пациентов с трисомией 21 имеют некоторую степень когнитивных нарушений, начиная от легкой (IQ 50-75) и заканчивая тяжелой формой (IQ 20-35). Все пациенты проявляют как моторные так и языковые задержки в детском периоде.
- Характерные черты лица
- Аномалии рук
- Врожденные пороки сердца – почти половина пациентов имеют врожденный пороки сердца, в том числе дефект межжелудочковой перегородки и дефекты атриовентрикулярного канала
Молекулярный анализ показывает, что область 21q22.1-q22.3, также известная как критическая область синдрома Дауна, содержит ген или гены, ответственные за развитие врожденных болезней сердца. Также, совсем недавно был обнаружен другой ген, DSCR1, который также находится в области 21q22.1-q22.2. Этот ген высоко выражается в головном мозге и сердце и на сегодня, он является основным кандидатом, ответственным за развитием умственной отсталости и сердечных дефектов.
Кольцевая 22 хромосома. Лечение
Лечение этого расстройства направлено только на контроль и избавление от конкретных симптомов и проявлений. Такое лечение может потребовать скоординированных усилий команды медицинских работников, таких как педиатров, хирургов и других специалистов.
Для некоторых пациентов, лечение может включать в себя хирургическое вмешательство для исправления некоторых черепно-лицевых или других физических аномалий, потенциально связанных с этим расстройством.
Типы и количество хирургических процедур, будут зависеть от тяжести анатомических нарушений, связанных с ними симптомов и других факторов.
Раннее вмешательство может быть важным для любого из таких пациентов.
Для детей также будут полезны физическая терапия, логопедия и / или другие медицинские, социальные и / или профессиональные направления. Генетическое консультирование также будет полезным для людей с кольцевой 22 хромосомой и их семей.
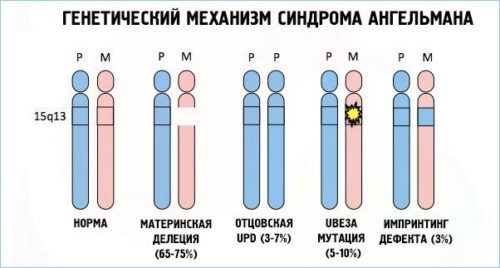
Синдром Тернера, или моносомия Х-хромосомы
Синдром Тернера – это генетическое заболевание, которое возникает в результате полного или частичного отсутствия одной Х-хромосомы в клетках организма. Исследования показывают, что более 90 процентов плодов с синдромом Тернера вымирают самопроизвольно. Заболевание вызывает около 10 процентов. самопроизвольные выкидыши. Одна из 2000–2500 девочек рождается с синдромом Тернера.
Заболевание проявляется:
- небольшой рост (максимум 145 сантиметров),
- коренастое телосложение,
- короткие нижние конечности,
- задержка полового созревания,
- нет волос на лобке,
- аменорея ,
- нарушение развития молочных желез,
- первичное бесплодие,
- многочисленные дисморфические признаки, такие как: припухлость ступней и кистей в форме подушки, избыток кожи на затылке, низко посаженные уши, опущенные веки, диагональные морщины, косоглазие, короткая и гибкая шея, готическое небо, низкая линия роста волос, пигментные родинки.
Около 30 процентов пациентов развиваются пороки сердца и аномалии почек. Что стоит подчеркнуть, психическое развитие девочек с синдромом Тернера правильное.
Причины генетической патологии
ДНКнуклеотидовбелок, какой-либо фермент или рецептор организмаВсе хромосомы в организме человека делятся на два вида:
- Аутосомы. Аутосомы – это хромосомные пары с 1 по 22. Они несут большой объем генетической информации и могут быть различных размеров. При синдроме Дауна у больных наблюдается утроение аутосомы под номером 21.
- Половые хромосомы. Половые хромосомы обозначаются цифрами Х и Y. Они предопределяют пол человека (XX – девочка, XY – мальчик). Условно эти хромосомы объединяют в 23-ю пару, хотя Х и Y не похожи друг на друга ни размером, ни формой, ни набором генов.
в последовательности нуклеотидовдля женщиндля мужчиндве хромосомы, составляющие пару, соединяются не в виде буквы Х, а в виде буквы VВ зависимости от характера хромосомной мутации различают следующие виды болезни:
- Полная трисомия 21. Полная трисомия 21 предполагает, что у ребенка в каждой клетке организма имеется целая дополнительная хромосома. Таким образом, общее количество ее копий – 3. Частота данного варианта составляет 90 – 95%. Эта форма является наиболее тяжелой. У пациента наблюдается избыток всех генов, закодированных в этой молекуле ДНК. Как правило, нарушения внутриутробного развития у них встречаются чаще, а умственная отсталость более выражена. Полная трисомия возникает, если один из родителей передает ребенку не одну, а две хромосомы 21. Тогда при слиянии с третьей 21-й хромосомой (от второго родителя) возникает трисомия. Зигота (первая клетка, из которой возникает зародыш) уже содержит дефект. Дальнейшее ее деление объясняет, что все дочерние клетки будут похожи на нее.
- Мозаичная форма. При мозаичной форме механизм появления хромосомного дефекта несколько иной. Обе родительские гаметы (половые клетки) имели нормальное количество хромосом. После их слияния образовалась нормальная зигота с кариотипом 46, ХХ или 46, XY. В процессе деления этой первоначальной клетки ДНК распределилось неправильно. Часть клеток организма получилась с нормальным кариотипом, а часть – с кариотипом синдрома Дауна. Такая аномалия встречается довольно редко (3 – 5% случаев данного заболевания). Прогноз при ней лучше, так как здоровые клетки отчасти компенсируют генетический дефект. Ребенок все равно родится с синдромом Дауна и видимым отставанием в развитии. Однако выживаемость таких детей значительно выше. У них редко встречаются тяжелые пороки развития внутренних органов, несовместимые с жизнью.
- Семейный синдром Дауна. Семейный синдром Дауна – это весьма редкий генетический дефект (менее 2% случаев). При нем один из родителей имеет небольшие отклонения. Часть хромосомы 21 (а именно, критический участок) прикрепляется к другой хромосоме (обычно к 14-й). Таким образом, на 14 хромосоме содержится больше генетической информации, чем должно быть в норме. У человека при этом обычно нет видимых изменений (симптомов синдрома Дауна). Однако все половые гаметы, которые производит его организм, содержат этот дополнительный участок хромосомы 21. Очень высока вероятность, что в процессе образования зиготы такая гамета вызовет появление дополнительной 21-й хромосомы. Таким образом, дети у человека с подобным дефектом часто рождаются с синдромом Дауна. Из-за этой аномалии, передающейся потомству, данную форму болезни назвали семейной.
- Частичная трисомия 21. При частичной трисомии 21 у пациента обнаруживается не вся дополнительная хромосома, а лишь ее фрагмент с критическим участком. Из-за этого у ребенка развивается синдром Дауна в более легкой форме (однако все основные симптомы все равно присутствуют). Механизм такого дефекта чем-то похож на семейную форму болезни, но синдром не будет передаваться по наследству. Встречается этот вариант болезни очень редко.
На образование аномальных гамет могут повлиять следующие факторы:
- экологическая обстановка;
- некоторые медикаменты;
- курение;
- алкоголизм;
- радиация;
- некоторые заболевания половой сферы.
зачать ребенкаВероятность рождения ребенка в зависимости от возраста матери выглядит следующим образом:
- 0,064% для женщин, рожающих в возрасте 20 – 24 лет;
- 0,1% — для женщин в возрасте 25 – 30 лет;
- 0,17% — для женщин в возрасте 31 – 35 лет;
- 0,47% – для женщин 36 – 40 лет;
- 0,78% — для женщин 41 – 45 лет;
- до 5,25% — у женщин старше 45 лет (синдром Дауна – у каждого двадцатого ребенка).
Трисомия по Х хромосоме. Причины
Трисомии по Х хромосоме представляют собой хромосомные аномалии, которые характеризуются наличием дополнительной Х-хромосомы.
Несмотря на то, что трисомия X является генетическим нарушением, она не наследуется. Наличие дополнительной Х-хромосомы является следствием ошибки при расхождении хромосом. Эти ошибки возникают случайно и без видимой причины (спорадически). В большинстве случаев, дополнительная Х-хромосома имеет материнское происхождение.
Примерно в 20 процентах случаев, нерасхождение происходит после зачатия. Исследователи считают, что симптомы и физические особенности этого расстройства связаны с сверхэкспрессией генов.
Патофизиология
Кариотип (хромосомный набор) человека с синдромом Дауна. В 21-й паре три хромосомы вместо двух
Синдром Дауна — хромосомная патология, характеризующаяся наличием дополнительных копий генетического материала 21-й хромосомы, либо целой хромосомы (трисомия), либо её участков (например, за счёт транслокации). Последствия от наличия дополнительной копии сильно различаются в зависимости от количества дополнительного генетического материала, генетического окружения и чистой случайности. Синдром Дауна встречается как у людей, так и у других видов (например, был обнаружен у обезьян и мышей). В 2005 году британские исследователи получили анеуплоидных трансгенных мышей с наличием 21-й человеческой хромосомы в дополнение к стандартному набору мышей. Нормальный человеческий кариотип содержит 46 хромосом и обозначается 46,XY у мужчин и 46,XX у женщин, в то время как у носителей синдрома Дауна с трисомией по 21-й хромосоме кариотип содержит 47 хромосом.
Трисомия
Трисомия — это наличие трёх гомологичных хромосом вместо пары в норме.
Риск рождения ребёнка с синдромом Дауна и другими численными хромосомными аномалиями растёт с возрастом матери. Точная причина этого неизвестна, но, по-видимому, она связана с возрастом яйцеклеток матери.
Трисомия происходит из-за нерасхождения хромосом во время мейоза, в результате чего возникает гамета с 24 хромосомами. При слиянии с нормальной гаметой противоположного пола образуется зигота с 47 хромосомами, а не 46-ю, как без трисомии.
Трисомия 21-й хромосомы в 95 % случаев является причиной возникновения синдрома Дауна, и в 88 % случаев из-за нерасхождения материнских гамет и в 8 % — мужских.
Мозаицизм
Трисомия обычно вызвана нерасхождением хромосом при формировании половых клеток родителя (гамет), в этом случае все клетки организма ребёнка будут нести аномалию. При мозаицизме же нерасхождение возникает в клетке зародыша на ранних стадиях его развития, в результате чего нарушение кариотипа затрагивает только некоторые ткани и органы. Данный вариант развития синдрома Дауна называется «мозаичный синдром Дауна» (46, XX/47, XX, 21). Данная форма синдрома является, как правило, более лёгкой (в зависимости от обширности изменённых тканей и их расположения в организме), однако более трудна для пренатальной диагностики.
По данному типу синдром появляется в 1—2 % случаев.
Робертсоновские транслокации
Дополнительный материал 21-й хромосомы, вызывающий синдром Дауна, может появиться вследствие наличия робертсоновской транслокации в кариотипе одного из родителей. В данном случае длинное плечо 21-й хромосомы прикреплено к плечу другой хромосомы (чаще всего 14-й ). Фенотип у человека с робертсоновскими транслокациями соответствует норме. Во время репродукции нормальный мейоз повышает шанс на трисомию 21-й хромосомы и рождения ребёнка с синдромом Дауна. Транслокации с синдромом Дауна часто называют семейный синдром Дауна. Эта форма не зависит от возраста матери. Данный тип появления синдрома занимает 2—3 % от всех случаев.
Дупликация части хромосомы 21
Очень редко участки 21-й хромосомы могут быть удвоены в результате хромосомной перестройки. При этом возникают дополнительные копии некоторых, но не всех генов из 21-й хромосомы. Если продублируются фрагменты, обусловливающие физические и психологические проявления синдрома Дауна, то ребёнок родится с этим синдромом. Такие хромосомные перестройки происходят крайне редко, и не существует оценки периодичности данного явления.
Формы синдрома Дауна
Примерно в 91 % случаев возникает ненаследственный вариант синдрома — простая полная трисомия 21 хромосомы, обусловленная нерасхождением хромосом во время мейоза. Примерно у 5 % людей с синдромом Дауна наблюдается мозаицизм (не все клетки содержат лишнюю хромосому). В остальных случаях синдром вызван спорадической или наследуемой транслокацией 21-й хромосомы. Как правило, такие транслокации возникают в результате слияния центромеры 21-й хромосомы и другой акроцентрической хромосомы. Фенотип больных определяется трисомией 21q22. Повторный риск рождения ребёнка с синдромом Дауна у родителей с нормальным кариотипом составляет около 1 % при обычной трисомии у ребёнка.
Информация об этих редких формах значима для родителей, так как риск рождения других детей с синдромом Дауна различен при разных формах. Тем не менее, для понимания развития детей эти различия не так важны. Хотя профессионалы склонны считать, что дети с мозаичной формой синдрома Дауна отстают в своём развитии меньше детей с другими формами этого синдрома, достаточно убедительных сравнительных исследований на эту тему пока нет.
Лечение
Лечение синдрома тройного Х зависит от возраста представления заболевания, его тяжести и симптомов.
Дети
Если новорожденному диагностирован синдром трисомии Х, ребенок должен оцениваться следующим образом:
- Первые 4 месяца: оценка развития мышечного тонуса и силы.
- До 12 месяцев: оценка языка, речи.
- В дошкольном возрасте: предварительная оценка ранних признаков проблем чтения.
- Для детей с синдромом тройного Х необходимо оценить функцию почек и сердца.
Детям с синдромом тройного Х, ранняя оценка и вмешательство дают отличный результат. Речевая, развивающая терапия, физиотерапия, консультирование являются ключевыми мерами вмешательства, когда это необходимо.
Лечение тревоги и СДВГ имеет важное значение при обнаружении
Молодые девушки
Для девочек с синдромом тройного Х подростковый возраст может быть сложной фазой жизни. Для них требуется короткий период консультирования.
Женщины
Для женщин с бесплодием и нарушениями менструального цикла требуется тщательная проверка наличия первичной овариальной недостаточности.
Диагностика трисомии 21
Хорошее здоровье родителей и благоприятно протекающая беременность – не гарантия того, что ребенок будет здоров. Существует такое понятие как базовый риск возникновения патологий. Под этим термином понимают пропорциональное отношения числа беременных женщин с одинаковыми характеристиками к количеству случаев заболевания трисомией 21
Для выявления хромосомных аномалий важно провести необходимую диагностику (скрининг) уже на ранних сроках беременности
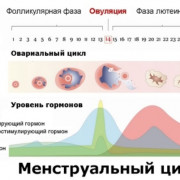
Уже в 1 триместре беременности женщины есть возможность посчитать индивидуальный риск возникновения хромосомных патологий у плода. Для этого необходимо провести ряд исследований, которые включают в себя, прежде всего, ультразвуковое исследование (УЗИ) и биохимический анализ крови.
Это одно из наиболее универсальных и безопасных обследований для диагностики трисомии. Первое УЗИ обычно приходится на 12 неделю беременности
Существуют определенные маркеры, на которые обращает внимание врач во время первого УЗИ и которые могут сигнализировать о наличии у плода отклонений:
- утолщение зоны воротникового пространства;
- отсутствие носовой кости;
- отставание роста и веса плода от нормы на 8 – 10%.
На УЗИ второго триместра специалист обращает внимание на наличие следующих признаков болезни:
- брахицефалическая форма головы (короткоголовость);
- увеличенный объем сердечных желудочков;
- киста в задней черепной ямке;
- недоразвитость костей лицевых структур;
- дополнительная складка на шее;
- непроходимость кишечника;
- пороки сердца;
- короткие трубчатые кости конечностей;
- аномалии развития пальцев;
- гидронефроз почек.
Согласно статистике, при наличии 3 – 4 из указанных признаков вероятность подтверждения диагноза трисомия 21 будет составлять 15 – 25%
Стоит принять во внимание, что ни один врач не будет ставить диагноз исключительно по данным УЗИ. Для составления полной картины необходимо провести и другие исследования, в том числе, биохимический анализ крови. Исследование крови матери
Исследование крови матери
Сывороточными маркерами называют вещества, которые возникают в крови женщины на разных сроках беременности. Было установлено, что концентрация этих маркеров заметно повышена или снижена относительно нормы у тех женщин, которые беременны ребенком с трисомией 21.
В первом триместре беременные сдают кровь на уровень хорионического гонадотропина человека (ХГЧ) и плазменного протеина А (PAPP-A). Во втором триместре таких маркеров будет три: ХГЧ, альфа-фетопротеин (АФП), эстриол свободный. Маркеры первого триместра целесообразно проверить с 10 по 14 неделю беременности, а во втором триместре сдать анализ между 16 и 18 неделями. Полученные показатели будут оцениваться относительно норм, предусмотренных для конкретной недели беременности.
Результаты УЗИ и биохимического скрининга всегда оцениваются в совокупности. Для вычисления индивидуального риска по трисомии 21 учитываются:
- данные УЗИ на сроке 11 – 13 недель;
- анализ крови на сывороточные маркеры;
- индивидуальные особенности беременной (возраст, вредные привычки, хронические заболевания).
Эти показатели обрабатываются компьютерной программой, которая и вычисляет вероятность того, что у плода могут быть отклонения. Например, результат скрининга 35-летней беременной — 1:95. Такие цифры говорят о высоком риске и о необходимости прибегнуть к дополнительным видам обследования. Для подтверждения или опровержения диагноза врачи направляют женщин из группы риска на инвазивные обследования. В зависимости от срока беременности это может быть: биопсия хориона, амниоцентез или кордоцентез.
Каждый из этих методов предполагает хирургическое вмешательство — пункцию брюшной стенки матери с целью забора материала, который содержит информацию о ДНК плода (ворсины хориона, околоплодная жидкость, пуповинная кровь). Методы эти очень точны (около 99%), но не совсем безопасны. В ряде случаев они могут спровоцировать выкидыш (вероятность около 1,5%).
В арсенале у современной медицины среди высокоточных способов пренатальной диагностики есть и безопасные методы, которые подразумевают лишь взятие венозной крови матери. Таким методом является неинвазивный пренатальный ДНК тест, который эффективен уже с 9 недели беременности и способен выявить широкий спектр хромосомных патологий, одна из которых как раз трисомия 21. Подробная расшифровка теста предоставляется будущим родителям в течение 14 дней с момента сдачи анализа.
Своевременное выявление синдрома Дауна позволяет семейной паре принять ответственное решение относительно того, готовы ли они к рождению больного ребенка и будет ли сохранена беременность.