Синдром ангельмана: клинические проявления и подходы к терапии
Содержание:
- Примечания
- Синдром Ангельмана диагностика
- Подготовка к назначению
- Симптомы и признаки заболевания
- Лечение синдрома Ангельмана
- Формы
- Диагностика
- Формы
- Лечение синдрома Ангельмана
- Группа риска
- Общие сведения
- Причины возникновения синдрома
- Симптомы
- Стадии болезни
- Когда мир придуманный желаннее реального
- Синдром Ангельмана – лечение
- Что такое синдром Ларона?
- Съёмочная группа
- Признаки и симптомы
Примечания
- Малая энциклопедия врача-эндокринолога / Под ред. А. С. Ефимова. — 1-е изд. — К.: Медкнига, ДСГ Лтд, Киев, 2007. — С. 317-318. — 360 с. — («Библиотечка практикующего врача»). — 5000 экз. — ISBN 966-7013-23-5.
- Whonamedit — Laron’s syndrome
- Laron Z, Pertzelan A, Mannheimer S (1966). «Genetic pituitary dwarfism with high serum concentration of growth hormone—a new inborn error of metabolism?». Isr. J. Med. Sci.2 (2): 152–5. PMID 5916640.
- Laron Z (2004). «Laron syndrome (primary growth hormone resistance or insensitivity): the personal experience 1958-2003». J. Clin. Endocrinol. Metab.89 (3): 1031–44. DOI:10.1210/jc.2003-031033. PMID 15001582.
- Galli-Tsinopoulou, A., Nousia-Arvanitakis, S., Tsinopoulos, I., Bechlivanides, C., Shevah, O. and Laron, Z. Laron syndrome. First report from Greece // Hormones. — 2003. — Vol. 2, № 2. — P. 120—124.
- Arlan L. Rosenbloom. Growth Hormone Insensitivity Syndrome // Pediatric Endocrinology: A Practical Clinical Guide / S. Radovick, M. H. MacGillivray (Eds.). — Humana Press, 2003. — P. 43. — ISBN 978-0-89603-946-9.
- ↑ 12345 Симптомы и синдромы в эндокринологии / Под ред. Ю. И. Караченцева. — 1-е изд. — Х.: ООО «С.А.М.», Харьков, 2006. — С. 91-92. — 227 с. — (Справочное пособие). — 1000 экз. — ISBN 978-966-8591-14-3.
- ↑ 12 Science Translational Medicine Vd. 3, issue 70
Синдром Ангельмана диагностика
Это заболевание диагностируется после генетического анализа пятнадцатой хромосомы. Диагностику назначают новорожденным с гипотонусом (пониженным мышечным тонусом), при отставании в развитии речи, а также общей моторики
Родителям следует обратить внимание на специфическое выражение лица, мелкий тремор, хаотические, а также порывистые движения конечностей, негнущиеся ноги во время походки и очень частый смех
Методы анализа включают флуоресцентную гибридизацию in situ, метилирование ДНК области 15q11- q13, а также анализ прямой мутации в гене UBE3A и анализ мутации импринтингового центра.
Существует отдельная группа людей, имеющая все признаки синдрома Ангельмана, однако при этом результаты всех вышеописанных анализов остаются в норме.
Подготовка к назначению
Позвоните своему врачу, если ваш ребенок или ребенок не достигли ожидаемых этапов развития или имеют другие симптомы или симптомы, общие для синдрома Ангельмана. Затем ваш врач может обратиться к врачу, который специализируется на состояниях, которые влияют на мозг и нервную систему (невролог).
Вот некоторая информация, которая поможет вам подготовиться к встрече.
Что ты можешь сделать
- Запишите признаки или симптомы, которые вы заметили у своего ребенка, и как долго.
- Принесите детские книги и другие записи о развитии вашего ребенка к назначению. Фотографии и видеозаписи могут быть полезны.
- Перечислите основную медицинскую информацию вашего ребенка,включая другие условия, в которых лечится ваш ребенок, а также названия лекарств, витаминов или добавок, которые он или она принимает.
- Попросите члена семьи или друга присоединиться к вам для назначения вашего ребенка. Если врач вашего ребенка упоминает о возможности развития расстройства, вам может быть трудно сосредоточиться на чем-либо, о чем говорит врач. Возьмите кого-нибудь, кто может предложить эмоциональную поддержку и поможет вам запомнить эту информацию.
- Запишите вопросы, чтобы спросить своего врача.
Вопросы, задаваемые врачом вашего ребенка, включают:
- Что может вызвать признаки и симптомы моего ребенка?
- Существуют ли другие возможные причины этих признаков и симптомов?
- Какие тесты нужен моему ребенку?
- Должен ли мой ребенок видеть специалиста?
Вопросы, задаваемые специалистом, включают:
- У моего ребенка синдром Ангельмана?
- Каковы возможные осложнения этого состояния?
- Какие методы лечения доступны?
- Какое лечение вы рекомендуете?
- Какова долгосрочная перспектива для моего ребенка?
- Должен ли мой ребенок или я пройти тестирование на генетические мутации, связанные с этим заболеванием?
- Какие другие специалисты должен видеть мой ребенок?
- Как я могу найти другие семьи, которые справляются с синдромом Ангельмана?
Не стесняйтесь задавать и другие вопросы.
Что ожидать от вашего врача
Врач, который видит вашего ребенка в возможном синдроме Англмана, скорее всего, задаст вам ряд вопросов, таких как:
- Каковы признаки и симптомы вашего ребенка и когда вы их заметили?
- У вашего ребенка проблемы с кормлением?
- Ваш ребенок достигает ожидаемых, связанных с возрастом физических вех?
- Вы заметили проблемы с балансом, координацией или движением?
- Ваш ребенок смеется, улыбается или выражает волнение чаще, чем его или ее сверстники?
- Ваш ребенок выражает волнение с необычным физическим поведением, таким как ручка?
- Сообщает ли ваш ребенок устно?
- Насколько хорошо спит ваш ребенок?
- У вашего ребенка были судороги? Если да, то как часто?
- Были ли у кого-либо из родственников первой степени вашего ребенка, например родителя или брата, был диагностирован синдром Ангельмана?
Поделиться ссылкой:
- Нажмите, чтобы поделиться на Twitter (Открывается в новом окне)
- Нажмите здесь, чтобы поделиться контентом на Facebook. (Открывается в новом окне)
- Нажмите, чтобы поделиться в Telegram (Открывается в новом окне)
Нравится
Симптомы и признаки заболевания
Первые признаки, на которые могут обратить внимания родители в младенческом возрасте – это задержка речевого и психомоторного развития. При этом невербальные навыки могут быть развиты намного лучше, чем вербальные. Видео с такими детьми можно найти в интернете.
Основные симптомы болезни:
- Очень слабый набор веса;
- Хаотичные движения рук и ног вплоть до тремора;
- Повышенная гиперактивность;
- Трудности с обучением и навыками;
- Возможное деформирование черт лица;
- Косоглазие;
- Приступы эпилепсии.
Вышеуказанные признаки не гарантируют наличие заболевания, а лишь указывают
При совпадениях большинства из них важно обратиться к генетику и сдать анализ на кариотип
Кариотип – это совокупность всех признаков (количество, форма, размер) в полном наборе хромосом человека.
Проявления некоторых признаков синдрома может изменяться по мере взросления. Наиболее ярким признаком в любом возрасте является сочетание счастливого поведения и беспричинного смеха.
Лечение синдрома Ангельмана
Хромосомные нарушения, лежащие в основе синдрома, устранить невозможно. Пациентам назначается симптоматическое лечение, психолого-педагогическая коррекция, реабилитационные мероприятия. Для уменьшения частоты эпилептических припадков используются антиконвульсанты, для нормализации сна – мелатонин. Занятия лечебной физической культурой и сеансы массаж направлены на развитие мелкой моторики и скоординированной походки, устранение сколиоза. Для улучшения коммуникативных навыков детей обучают языку жестов, вовлекают в групповые занятия, организуют сеансы поведенческой терапии, позволяющей освоить правила взаимодействия в обществе.
Продолжается поиск способов эффективного лечения синдрома. Проводится тестовое применение препаратов на генетически модифицированных мышах. Результаты доказывают, что ингибиторы топоизомеразы способны активировать материнский ген UBE3A. На данном этапе выполняются контрольные исследования, определяется безопасность и риски терапии, но информации пока недостаточно для перенесения экспериментов на группы людей.
На сегодняшний день еще не изобретен чудо-препарат, который бы помог победить это генетическое заболевание. Однако болезнь Ангельмана предполагает симптоматическую терапию, благодаря которой облегчается состояние пациента. При этом прописывается медикаментозное и немедикаментозное лечение. Синдром Петрушки у детей предусматривает такую терапию:
- Назначаются антиконвульсанты. Чаще прописывают Клоназепам, Конвулекс, Ламотриджин. Такие лекарственные средства сводят к минимуму частоту и интенсивность эпиприступов.
- Прописывают витаминотерапию (элементы групп B, C, D и E). Такое лечение укрепляет иммунную систему организма. Однако оно уменьшает эффективность противоэпилептических средств, поэтому все назначения должен делать доктор.
- Прописывается терапия, направленная на устранение проблем с пищеварительным трактом. Такое лечение предусматривает прием слабительных препаратов (Фитолакса или Сенаде) и пробиотиков (Хилак форте, Бифиформа).
- Назначают снотворные. Чаще пациентам прописывают Дифенгидрамин или Мелатонин.
- Показана гормональная терапия. Такое лечение направлено на коррекцию поведения пациентов. Например, гормон секретин улучшает процесс пищеварения, а окситоцин – увеличивают познавательные способности и память.
- В борьбе с проблемами суставов помогут физиопроцедуры. Назначаться могут парафиновые аппликации, массаж, магнитотерапия, электрофорез, аквагимнастика.
При этом заболевании показана поведенческая терапия, занятия с логопедом, психологом и дефектологом. У детей, у которых диагностирован синдром счастливой куклы, отмечаются серьезные нарушения речи. Некоторые из них имеют ограниченный словарный запас, а другим даже сложно воспроизводить отдельные звуки.
Занятия с такими детками должны быть непродолжительными (не более 30 минут), но регулярными: желательно, чтобы они проводились каждый день. По мнению специалистов, начинать можно с рисования пальчиковыми красками. Во время таких занятий ребенок успокаивается, знакомится с цветовой гаммой и развивает мелкую моторику.
Поскольку дети с синдромом Ангельмана лучше воспринимают информацию, когда она визуализирована, желательно при их обучении использовать карточки и другие наглядные пособия. Например, чтобы объяснить такому ребенку понятия «большой»» и «маленький», можно воспользоваться разборной матрешкой. При первом знакомстве малыша с таким наглядным пособием взрослый должен сам открыть игрушку и каждое свое действие прокомментировать. Затем можно попросить ребенка проделать то же самое. Постепенно задача усложняется: например, малыш должен показывать, где большая матрешка.
Формы
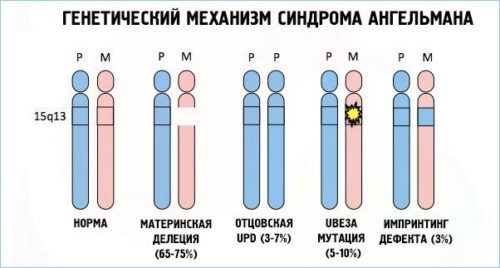
Синдром Ангельмана связан с четырьмя вариантами генетических мутаций:
- Вновь возникшей хромосомной мутацией, которая связана с потерей участка хромосомы в локусе 15 q11 — q13 . Данная мутация – причина около 80 % всех случаев заболевания.
- Одноотцовской дисомией, которая связана с потерей материнского локуса (отсутствие генетического материала матери). Данный вариант встречается редко (около 5% всех случаев).
- Дефектом ряда генов, подверженных геномному импринтингу (ГИ). Данные дефекты возникают у 2-4% больных в результате непосредственного нарушения импринтинга (различия в преобразовании информации гена в белок или РНК, которые зависят от происхождения гена). Чаще всего возникает в результате утраты центра регуляции ГИ. Дефекты ГИ без утраты центра регуляции являются результатом спонтанной мутации, повторение которой – большая редкость.
- Спонтанной мутацией материнской копии, которая вызывает отсутствие преобразования в мозге копии гена UBE3A. Данный ген кодирует деятельность убиквитинлигазы (фермент, участвующий в сложном процессе распада белков). Дефицит данного фермента относится к молекулярным механизмам синдрома.
Установить форму заболевания у 7-9 % в настоящее время не представляется возможным.
Диагностика
Постановка диагноза допускается еще при нахождении ребенка в утробе матери посредством неинвазивного или инвазивного метода.
Первый метод безопасен, исключается проникновение в матку, тогда как при инвазивном методе для получения результатов необходимо проникнуть в матку и взять околоплодную жидкость, что нельзя назвать совершенно безопасной мерой.
Для неинвазивного метода достаточно взять кровь матери на анализ состава ДНК младенца. Данная методика позволяет также диагностировать синдром Прадера-Вилли, Патау, Эдвардса и синдром Дауна.
Все упомянутые патологии неизлечимы и могут протекать в разных формах.
После рождения ребенка в обязанности родителей будет входить тщательное отслеживание поведения, выражения лица и эмоций, движений конечностями.
Проведение полной диагностики потребуется при малейших подозрениях в наличии отклонений. Синдром Ангельмана диагностируется с максимальной достоверностью чаще всего уже в 3-7 лет, когда важные для диагностики симптомы уже ярко выражены.
Характер повреждения напрямую влияет скорость прогрессирования болезни, поэтому жизнь одних пациентов может быть достаточно самостоятельной, а другие даже разговаривать не смогут.
Посредством генетических исследований удастся определить аномалии, которые выступают в качестве провоцирующего фактора. Только в комплексе с учетом признаков и результатов обследований можно достоверно поставить диагноз. В 20% случаев точно установить причину так и не удается, и чтобы хоть как то продвинуться в решении проблемы специалисты проводят много исследований.
Формы
Синдром Ангельмана связан с четырьмя вариантами генетических мутаций:
- Вновь возникшей хромосомной мутацией, которая связана с потерей участка хромосомы в локусе 15 q11 — q13 . Данная мутация – причина около 80 % всех случаев заболевания.
- Одноотцовской дисомией, которая связана с потерей материнского локуса (отсутствие генетического материала матери). Данный вариант встречается редко (около 5% всех случаев).
- Дефектом ряда генов, подверженных геномному импринтингу (ГИ). Данные дефекты возникают у 2-4% больных в результате непосредственного нарушения импринтинга (различия в преобразовании информации гена в белок или РНК, которые зависят от происхождения гена). Чаще всего возникает в результате утраты центра регуляции ГИ. Дефекты ГИ без утраты центра регуляции являются результатом спонтанной мутации, повторение которой – большая редкость.
- Спонтанной мутацией материнской копии, которая вызывает отсутствие преобразования в мозге копии гена UBE3A. Данный ген кодирует деятельность убиквитинлигазы (фермент, участвующий в сложном процессе распада белков). Дефицит данного фермента относится к молекулярным механизмам синдрома.
Установить форму заболевания у 7-9 % в настоящее время не представляется возможным.
Лечение синдрома Ангельмана
Хромосомные нарушения, лежащие в основе синдрома, устранить невозможно. Пациентам назначается симптоматическое лечение, психолого-педагогическая коррекция, реабилитационные мероприятия. Для уменьшения частоты эпилептических припадков используются антиконвульсанты, для нормализации сна – мелатонин. Занятия лечебной физической культурой и сеансы массаж направлены на развитие мелкой моторики и скоординированной походки, устранение сколиоза. Для улучшения коммуникативных навыков детей обучают языку жестов, вовлекают в групповые занятия, организуют сеансы поведенческой терапии, позволяющей освоить правила взаимодействия в обществе.
Продолжается поиск способов эффективного лечения синдрома. Проводится тестовое применение препаратов на генетически модифицированных мышах. Результаты доказывают, что ингибиторы топоизомеразы способны активировать материнский ген UBE3A. На данном этапе выполняются контрольные исследования, определяется безопасность и риски терапии, но информации пока недостаточно для перенесения экспериментов на группы людей.
Группа риска
В качестве факторов риска и провоцирующих элементов стоит рассмотреть родительские хромосомные аномалии.
- Кольцевая хромосома – удаление генетического материала с концов хромосомы и соединение новообразованных концов в кольцо.
- Дупликация — частичное повторение хромосом и соответствующее появление лишнего генетического материала.
- Транслокация – присоединение к хромосоме элемента другой хромосомы.
- Делеция предполагает отсутствие одного из секторов хромосомы.
- Микроделеция является следствием перестройки Y-хромосомы и обмена секторов между половыми хромосомами при мейозе. Немного хромосом при этом отсутствует, чаще всего одного из генов тоже нет.
- Инверсия – разворот на 180 градусов одного из участков хромосомы, порядок расположения генов обратный, а часть хромосомы опущена.
- Трисомии хромосом обусловлена наличием одной или больше лишних хромосом в наборе. Причиной такого генетического дефекта является нерасхождение хромосом при делении.
Общие сведения
Фото мальчика с синдромом Ангельмана:
Синдром Ангельмана представляет собой патологию, которая возникает в результате генетических мутаций, в частности, из-за потери фрагмента 15 хромосомы.
Генетическая мутация может проявляться в результате нарушений хромосомного наследования, когда ребенок наследует 2 отцовские хромосомы, вместо 1 отцовской и 1 материнской, при перемещении фрагмента хромосомы, при изменении того или иного ее фрагмента.
Заболевание не всегда является переданным по наследству, нередко случается так, что больной ребенок появился у родителей, не страдающих какими-либо генетическими отклонениями.
Таким образом, можно сделать вывод, что мутационные процессы, приводящие к возникновению недуга, носят спонтанный характер, и предсказать рождение ребенка с подобными отклонениями весьма проблематично.
Причины возникновения синдрома
Причины возникновения продолжают изучаться. Особая роль отводится генетической предрасположенности, а также мутации в геноме, связанного с X хромосомой.
Мутация в гене, кодирующего метил-СрG-связывающий гликопротеин. приводит к нарушению правильного развития деятельности головного мозга в эмбриональном периоде развития.
Если ген не изменен, то мозг и его отделы развиваются без патологии. Если произошла мутация, происходит поражение X- хромосомы. Так как у женщин их две, то одна X-хромосома – нормальная, а вторая – дефектная.
С одной нормальной X-хромосомой девочкам удается появится на свет. Так как у мальчиков набор хромосом состоит из X и Y- хромосом, то их рождение с синдромом Ретта теоретически невозможна. Они погибают во внутриутробном периоде. Но существуют исключения.
При полисомии XXY хромосомах, рождение мальчиков возможно (синдром Клайнфельтера).
Симптомы
Для заболевания характерно отсутствие:
- отклонений в течение перинатального периода;
- выраженных пороков развития при рождении.
Окружность головы новорожденного не отклоняется от нормы.
Основные характерные симптомы синдрома Ангельмана проявляются в возрасте 6 -12 месяцев. В этом возрасте у больных можно выявить очевидную задержку развития при отсутствии прогрессирующей утраты приобретенных навыков. Результаты лабораторных тестов не отклоняются от нормы, обследования при помощи МРТ или КТ не показывает наличие структурных изменений в головном мозге. Возможна умеренная кортикальная атрофия и проявление избирательного повреждения миелиновой оболочки (демиелинизация).
К обязательным симптомам, которые встречаются во всех случаях заболевания, относят:
Выраженную задержку психического развития.
Нарушения речи. Возможно ее полное отсутствие или минимальный (около 6 лексических единиц) запас слов.
Расстройство моторных функций
Проявляется в нарушении координации движений (атаксия), равновесия, мелком треморе и хаотическом движении рук и ног.
Нарушения поведения (гиперактивность, практически постоянная улыбка и частый смех без видимой причины, пониженная способность концентрировать внимание).
Способность к невербальному общению сохранена, больные понимают больше, чем способны выразить.
Присутствуют стереотипии (повторяющиеся взмахи руками, кручения кистями и частое хлопанье в ладоши, не характерные для здоровых людей).
У 80% больных наблюдаются:
- Задержка роста головы (при рождении окружность головы обычно меньше 32 см, а к году составляет 42 см.). Абсолютная или относительная микроцефалия развивается до двухлетнего возраста.
- Эпилепсия (по сложившемуся среди специалистов мнению это симптоматическая (вторичная) эпилепсия). На ЭЭГ фиксируется наличие большой амплитуды и низкой временной динамики волн.
У 20-80 % больных синдром Ангельмана сопровождается:
- Уплощением затылка.
- Нарушенным контролем за движениями языка, что вызывает трудности при приеме пищи. У новорожденных наблюдаются проблемы при грудном вскармливании, поэтому дети плохо набирают вес.
- Повышенной потребностью в жидкости (постоянное ощущение жажды).
- Обильным слюноотделением и высунутым языком.
- Страбизмом (косоглазием).
- Сколиозом.
- Наличием широкого рта с крупными редкими зубами.
- Усиленными сухожильными рефлексами.
- Выступающим вперед подбородком (прогнатия).
- Нарушениями сна.
Больные ходят на негнущихся ногах, создавая впечатление марионетки. Плечи при ходьбе могут быть приподнятыми, а руки – полусогнутыми.
При утрате части хромосомы (делеции) больные обычно светловолосы и светлоглазы, также наблюдается гипопигментация кожи.
В воде люди с синдромом Ангельмана чувствуют себя комфортнее, душные помещения и повышенную температуру переносят плохо.
Также может присутствовать диффузная гипотония мышц и пищеводный рефлюкс.
Стадии болезни
Выделяют 4 стадии заболевания:
- Первая стадия развивается в начале четвертого месяца жизни и может продолжаться до двух лет. Появляются отклонения физического развития ребенка, отсутствует интерес к происходящему, апатия, задержка роста головы, набора веса, низкий мышечный тонус и вялость.
- Вторая стадия развивается к году жизни, когда ребенок ползает, переворачивается, ходит, говорит. В течение года эти приобретенные навыки исчезают. Он не помнит, как разговаривать, ходить и т.д. У ребенка возникают стереотипные движения рук, напоминающие мытье под краном, нарушение дыхания во время отдыха и не только, расстройство координации движений. Также возникает судорожный синдром, напоминающий эпилептический припадок. Лечение эпилептического припадка не приводит к исчезновению судорог.
- Третья стадия чаще наблюдается в конце второго начале третьего годика. Этот этап еще называют стабилизацией. На этом этапе происходит регресс развития головного мозга, т.е развивается умственная отсталость, уменьшается количество эпилептиморфных припадков. Ребенок может находится в одном положении длительное время (находится «в себе»), но при этом нет беспокойства и криков.
- Четвертая стадия является самой тяжелой, развивается примерно к 5 -10 годам и характеризуется необратимыми дегенеративными процессами в центральной и периферической нервной системах, а также в суставах и позвоночном столбе. Развивается искривление позвоночника, ведущее в 3-4 степени сколиоза. Характерна кахексия (снижение веса ребенка), мышечная гипотония, уменьшение размеров головного мозга, боли в суставах и мышцах. Эпилептические припадки появляются очень редко.
Течение заболевания основывается на скорейшем установлении диагноза. Чем раньше был установлен диагноз, проведены лечебные и профилактические мероприятия, тем благоприятнее течение болезни. У разных детей течение патологии происходит по-разному. Если установление диагноза было выставлено в поздние сроки, велик риск развития тяжелейших осложнений, препятствующих нормальной жизни больного ребенка.
Когда мир придуманный желаннее реального
В фильме сказка переплетается с реальностью. И это отражение сложного внутреннего мира главного героя Пети, которого играет Евгений Миронов.
Петя – носитель звуко-зрительного сочетания векторов. Человек со звуковым и зрительным векторами – обладатель мощнейшего абстрактно-образного интеллекта. В искусстве это очень талантливые люди, способные придавать своим творениям необычайную глубину.
Петя – гениальный кукольник. Его куклы неординарны: они почти живые, каждая из них обладает своим характером. Петр живет в придуманном им мире кукол и не желает соприкасаться с миром реальным. Когда речь идет о семейной жизни, он даже говорит своему другу Борису: «Зачем они вообще нужны – эти дети? Это же банальщина. Мне интереснее заводить кукол». За таким восприятием мира стоит череда детских психологических травм.
В детстве Петя видит, как из окна выбрасывается рыжеволосая женщина. Это отпечатывается в детском сознании подсознательным ужасом – зрительные дети очень впечатлительны. Они рождаются со страхом смерти, их надо оберегать от подобных эпизодов. Их огромный эмоциональный потенциал нуждается в развитии, выведении наружу, в любовь и сострадание. У Пети нет таких условий. Он предоставлен сам себе.
Его родителям не до него. Они все время скандалят и кричат. Для ребенка со звуковым вектором, от природы обладающим очень чувствительным слухом, это настоящая травма.
В таких условиях звуковой ребенок все больше и больше отгораживается от мира, погружается внутрь себя, вплоть до аутизма. Однако Петя находит выход в игре в куклы. Зрение помогает звуку выжить в этом мире. Петр оживляет кукол, разговаривает с ними, создает вместе с ними параллельный мир, основанный на его богатой фантазии. Мир, который живет по законам, понятным для ребенка, и защищает его от грубого и несправедливого внешнего воздействия.
Детское спасительное увлечение перерастает в профессию, которая также полностью захватывает все его сознание и время. Однако есть одна ниточка, которая постоянно связывает его с реальностью и не дает полностью уйти в вымышленный мир кукол – это любовь к рыжеволосой Лизе.
Синдром Ангельмана – лечение
На сегодняшний день еще не изобретен чудо-препарат, который бы помог победить это генетическое заболевание. Однако болезнь Ангельмана предполагает симптоматическую терапию, благодаря которой облегчается состояние пациента. При этом прописывается медикаментозное и немедикаментозное лечение. Синдром Петрушки у детей предусматривает такую терапию:
- Назначаются антиконвульсанты. Чаще прописывают Клоназепам, Конвулекс, Ламотриджин. Такие лекарственные средства сводят к минимуму частоту и интенсивность эпиприступов.
- Прописывают витаминотерапию (элементы групп B, C, D и E). Такое лечение укрепляет иммунную систему организма. Однако оно уменьшает эффективность противоэпилептических средств, поэтому все назначения должен делать доктор.
- Прописывается терапия, направленная на устранение проблем с пищеварительным трактом. Такое лечение предусматривает прием слабительных препаратов (Фитолакса или Сенаде) и пробиотиков (Хилак форте, Бифиформа).
- Назначают снотворные. Чаще пациентам прописывают Дифенгидрамин или Мелатонин.
- Показана гормональная терапия. Такое лечение направлено на коррекцию поведения пациентов. Например, гормон секретин улучшает процесс пищеварения, а окситоцин – увеличивают познавательные способности и память.
- В борьбе с проблемами суставов помогут физиопроцедуры. Назначаться могут парафиновые аппликации, массаж, магнитотерапия, электрофорез, аквагимнастика.
Умственные упражнения для детей с синдромом Ангельмана
При этом заболевании показана поведенческая терапия, занятия с логопедом, психологом и дефектологом. У детей, у которых диагностирован синдром счастливой куклы, отмечаются серьезные нарушения речи. Некоторые из них имеют ограниченный словарный запас, а другим даже сложно воспроизводить отдельные звуки. Все это создает препятствия в общении с другими людьми. Для социализации таких пациентов используют особое обучение. Например, их учат языку жестов.
Занятия с такими детками должны быть непродолжительными (не более 30 минут), но регулярными: желательно, чтобы они проводились каждый день. По мнению специалистов, начинать можно с рисования пальчиковыми красками. Во время таких занятий ребенок успокаивается, знакомится с цветовой гаммой и развивает мелкую моторику. Свой первый рисунок он выполнит с подсказками взрослого, однако в будущем с увлечением начнет сам создавать шедевры. Как только малыш освоит технику рисования пальчиками, можно приступить к усвоению удержания кисти в руке.
Поскольку дети с синдромом Ангельмана лучше воспринимают информацию, когда она визуализирована, желательно при их обучении использовать карточки и другие наглядные пособия. Например, чтобы объяснить такому ребенку понятия «большой»» и «маленький», можно воспользоваться разборной матрешкой. При первом знакомстве малыша с таким наглядным пособием взрослый должен сам открыть игрушку и каждое свое действие прокомментировать. Затем можно попросить ребенка проделать то же самое. Постепенно задача усложняется: например, малыш должен показывать, где большая матрешка.
Что такое синдром Ларона?
Синдром Ларона — это группа крайне редких генетических нарушений, при которых организм не может использовать гормон роста (сокр. ГР), который он производит. Синдром Ларона может быть вызван мутациями в гене рецептора соматотропного гормона роста (СТГ) или мутациями в генах, участвующих в пути действия в клетке после того, как гормон роста связывается с его рецептором, предотвращая выработку инсулиноподобного фактора роста 1 (ИФР-1, IGF-1), вещества, ответственного за воздействие гормона роста на организм. Еще реже дети с делецией гена ГР, которых лечили рекомбинантным ГР, вырабатывают антитела, которые блокируют связывание гормона роста с его рецептором. Пострадавшие дети не могут нормально расти.
Дети с дефицитом СТГ, которых лечат ИФР-1 до наступления половой зрелости, имеют улучшенный рост, но, в отличие от детей с дефицитом ГР, получающих лечение рекомбинантным ГР, у них не восстанавливается нормальный рост. Лечение этих состояний эффективно только тогда, когда растущие кости еще открыты, т.е. до завершения подросткового периода. Нечувствительность к ИФР-1 из-за мутации рецептора ИФР-1 имитирует синдром Ларона, но приводит к менее выраженной недостаточности роста и в некоторой степени реагирует на лечение рекомбинантным ГР.
Синдром Ларона характеризуется низким ростом и задержкой роста костей, а также нормальным или высоким уровнем циркулирующего соматотропина. Другими распространенными симптомами являются задержка полового созревания, выпуклый лоб, низкий уровень сахара в крови в младенчестве и раннем детстве и ожирением во взрослом возрасте. За исключением крайне редкой формы синдрома, где ген ИФР-1 имеет дефекты, но развитие мозга нормальное, по-видимому, потому что ИФР-1 может быть получен в течение жизни плода без стимуляции СТГ в других условиях. Некоторые, но определенно не все, пациенты с менее редким состоянием дефицита рецептора ИФР-1 могут иметь легкое интеллектуальное нарушение.
Съёмочная группа
- Автор сценария: Алёна Алова по роману Дины Рубиной «Синдром Петрушки»
- Режиссёр-постановщик: Елена Хазанова
- Оператор-постановщик: Азиз Жамбакиев
- Художник-постановщик: Наталья Навоенко
- Композитор: Николя Рабеюс
- Санкт-Петербургский симфонический оркестр
- Хореограф: Раду Поклитару
- Директор картины: Надежда Попова
- Со-продюсеры: Ундине Филтер, Томас Крал, Пьер-Андре Тьебо совместно с Антонио Эксакустос, Йозеф Райдингер
- Продюсеры: Илья Гаврилов, Александр Новин, Дмитрий Аронин, Ася Темникова, Елена Бренькова, Анна Качко
- Художественный руководитель: Евгений Миронов
Признаки и симптомы
Существует широкий спектр симптомов в зависимости от вовлеченных генных мутаций (см. раздел «Причины»). Очень немногие люди с мутацией гена ИФР-1 имеют тяжелые нарушения умственного развития и нарушения внутриутробного развития с глухотой и микрогнатией. Дефицита СТГ приводит к серьезной недостаточности роста без вредного воздействия на внутриутробный рост или развитие мозга, а мутация STAT5b, ответственного за важный белок-активатор, оказывает аналогичные эффекты на рост, но также связана с серьезным нарушением иммунокомпетентности. IGFALS мутации, затрагивающие важную стабилизирующую составляющую циркулирующего ИФР-1, хотя и ассоциируется с очень низким уровнем циркулирующего ИФР, имеет лишь умеренное воздействие на рост.
Синдром Ларона характеризуется тяжелым, но пропорциональным невысоким ростом в результате нарушения роста, которое начинается при рождении. Наряду с задержкой роста, есть задержки в прорезывании зубов. Существует также диспропорция между ростом черепа и лица, седловым носом и глубоко посаженными глазами. Половое развитие умеренно задерживается у обоих полов. У женщин с этими расстройствами начало менструации обычно происходит в возрасте 16-19 лет. Руки и ноги меньше, чем обычно, пропорционально общему размеру тела. Также у взрослых больных может присутствовать высокий голос и ожирение, особенно у женщин.
Высокие уровни ГР в крови обнаруживаются у детей, но могут быть не очевидны без стимуляции у взрослых. Высокий процент молодых пациентов имеет низкий уровень сахара в крови (гипогликемия), который может быть связан с судорогами у некоторых очень маленьких детей. Недавно исследователи обнаружили, что популяция людей с дефицитом СТГ в Эквадоре (где была идентифицирована приблизительно 1/3 населения мира с дефектом рецепторов СТГ) имела отсутствие рака и диабета с молекулярными доказательствами защиты от изменений старения в их ДНК. Это может быть связано с защитным эффектом от низких уровней ИФР-1, а в случае отсутствия диабета, несмотря на ожирение, из-за отсутствия контррегуляторных эффектов гормона роста.