Мальчик стал сверхдобрым и доверчивым из-за синдрома вильямса
Содержание:
- Диагностика и лечение синдрома Вильямса
- Симптомы и признаки
- Лечение синдрома Ангельмана
- БРАТЬЯ И СЕСТРЫ
- КУДА ОБРАЩАТЬСЯ В ПОДМОСКОВЬЕ ПРИ ПОДОЗРЕНИИ НА ГЕНЕТИЧЕСКИЕ ОТКЛОНЕНИЯ?
- Лечение синдрома Вильямса — возможно ли?
- Методы лечения
- Что делать?
- Примечания
- Формы синдрома Элерса-Данлоса
- Симптомы синдрома Вильямса
- Отличительные признаки аномалии
- Факторы-провокаторы синдрома Вильямса
- ДИАГНОЗ
Диагностика и лечение синдрома Вильямса
Диагноз синдром Вильямса ставят на основании настоящего статуса больного, результатов биохимического анализа крови, кардиологических и молекулярно-генетических исследований. При осмотре обнаруживают характерный внешний вид («лицо эльфа»), нарушения формирования зубов и кариес, мышечную гипотонию с сопутствующими изменениями опорно-двигательного аппарата, признаки умственной неполноценности и отставания в физическом развитии. Картина биохимического анализа крови при синдроме Вильямса достаточно разнообразна, практически всегда при этом заболевании определяется гиперкальциемия, повышение уровня тиреотропного гормона, некоторое снижение уровня нейтрофилов. Как правило, изучение наследственного анамнеза больного не имеет смысла за исключением случаев наличия синдрома Вильямса у одного из родителей.
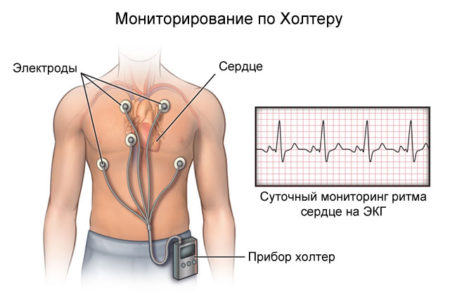
Кардиологические исследования (ЭхоКГ) могут выявлять врожденные пороки сердца, чаще всего – недостаточность аортального или митрального клапанов, надклапанные стенозы аорты и легочного ствола. У больных синдромом Вильямса старшего возраста может определяться утолщение стенок аорты и других магистральных сосудов, выраженная артериальная гипертензия. При осмотре косвенными признаками заболевания могут служить пупочные и паховые грыжи, сходящееся косоглазие, хрипловатый низкий голос. Самым достоверным диагностическим методом при синдроме Вильямса является молекулярно-генетический анализ, проводящийся врачом-генетиком. Для выявления отсутствия участка 7 хромосомы используют технику ДНК-микрочипа или флуоресцентной гибридизации (FISH-методика), возможна пренатальная диагностика.
Специфического лечения синдрома Вильямса не существует, применяют паллиативные мероприятия и методы психологической коррекции. Иногда требуется хирургическое вмешательство в раннем возрасте для исправления врожденных пороков сердца, которые в тяжелых случаях могут привести к летальному исходу. Для снижения выраженности артериальной гипертензии используют традиционные препараты, в основном из группы ингибиторов АПФ. Психокоррекционная работа при синдроме Вильямса позволяет больным овладеть речью, в некоторых случаях – чтением и письмом, уменьшает вероятность развития тревожности и синдрома навязчивых состояний. Немаловажную роль в снижении выраженности умственной отсталости играет дружелюбный эмоциональный фон в семье.
Прогноз и профилактика синдрома Вильямса
Прогноз выживаемости у страдающих синдромом Вильямса относительно благоприятный – наибольшую угрозу в раннем возрасте составляют врожденные пороки сердца, но при их слабой выраженности или своевременном устранении больные могут дожить до преклонного возраста. Продолжительность жизни обычно несколько ниже, чем у здоровых людей, что обусловлено стойким повышением артериального давления, нарушениями кальциевого и углеводного обменов. Хотя при синдроме Вильямса возможна частичная социальная адаптация, большинство больных нуждаются в тщательном уходе и контроле со стороны родственников. Возможность какой-либо трудовой деятельности при наличии этого заболевания полностью исключена. Профилактики синдрома Вильямса не существует, так как это заболевание обычно является результатом спонтанной мутации – возможно лишь пренатальное определение данной патологии.
Симптомы и признаки
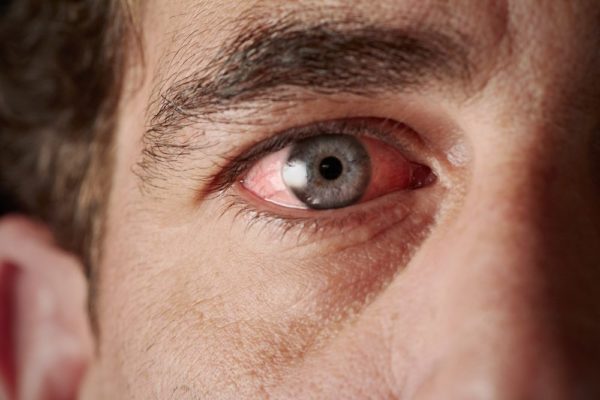
Ребенка с синдромом Вильямса можно распознать по характерной эльфийской внешности, низкому голосу, синеватому оттенку склер.
У многих детей присутствует плоскостопие, что влияет на походку: при ходьбе ребенок немного наклоняется вперед.
У большинства детей обнаруживаются следующие отклонения:
- Патологии сердечно-сосудистой системы. У них часто обнаруживается надклапанный аортальный стеноз, стенозы крупных артерий. Степень выраженности нарушения может варьироваться в зависимости от особенностей заболевания. Также присутствует клапанная недостаточность сердца и другие отклонения.
- Избыточная концентрация кальция. Наблюдается у большинства детей с синдромом Вильямса, часто исчезает самостоятельно в первые годы жизни, но может сохраняться десятилетиями (если это произошло, это нарушение постепенно усугубляет течение заболеваний внутренних органов, характерных для синдрома).
- Умственная отсталость. Выражена средне, уровень IQ находится в границах 40-80, в редких случаях поднимается практически до нормальных показателей. Максимально зафиксированный уровень IQ у человека с синдромом Уильямса — 110. Частично компенсируется особенностями поведения ребенка.
Первые слова ребенок говорит в 2-3 года, а способность соединять их в осмысленные фразы развивается еще позже — после 4 лет.
Нарушения в процессе набора веса. Младенцы с этой патологией медленно набирают вес, в дальнейшем отмечается умеренное отставание в физическом развитии. Их рост редко достигает 160 сантиметров.
Трудности с кормлением. Младенцы часто срыгивают, плохо берут грудь из-за нарушений в мышечной системе.
Болезни желудочно-кишечного тракта. Могут возникать выпячивания в кишечных стенках, которые становятся причиной хронических запоров, также часто наблюдается хронический гастрит типа С.
Нарушения гормонального фона. Если в процессе внутриутробного развития щитовидная железа не сформировалась, это приводит к возникновению гипотиреоза. Также люди с синдромом Вильямса склонны к развитию сахарного диабета.
Недоразвитость почек, которая приводит к возникновению мочекаменной болезни. Наблюдаются выпячивания в стенках мочевого пузыря.
Косоглазие, обычно сходящегося типа.
Повышенная слуховая чувствительность. Дети чувствительны к звукам, болезненно реагируют на те звуки, которые для обычных людей не создают дискомфорта.
Проблемы с мышечной системой и суставами. Мышечная система ослаблена, а суставы нестабильны, что приводит к прогрессированию нарушений в костных структурах. Отмечаются отклонения в моторике и координации.
Нарушения в костной системе. Характерно возникновения плоскостопия, различных искривлений позвоночного столба, деформации костей в зоне грудной клетки, вальгусное искривление нижних конечностей (колени соединяются).
Заболевания зубов. Отмечаются нарушения прикуса, риск развития кариеса и прочих проблем с зубами повышен.
Часть упомянутых нарушений могут отсутствовать у отдельно взятого ребенка с патологией, но ключевые нарушения, включая специфическую внешность и умственную отсталость, всегда присутствуют.
Также присутствуют характерные интеллектуальные и психологические особенности:
Дети с синдромом крайне дружелюбны, стремятся к общению, выглядят милыми и обаятельными, умеют хорошо разговаривать, чем привлекают к себе внимание. Вежливы, обходительны, улыбчивы
В первые годы жизни тревожно относятся к незнакомцам, но позднее эта проблема сглаживается.
Они практически не могут подмечать мимические особенности лиц других людей, говорящие о том, что человек относится к ним негативно. Это (как и чрезмерное дружелюбие) связано с отклонениями в развитии миндалевидного тела.
Им интересна музыка, они хорошо ее чувствуют и способны добиться успеха в этой сфере.
Несмотря на наличие интеллектуальных дефектов, подросшие дети имеют хороший словарный запас, но многие слова ими могут использоваться неправильно.
Склонны к перепадам настроения, тревожны, может наблюдаться недержание мочи.
Лечение синдрома Ангельмана
Хромосомные нарушения, лежащие в основе синдрома, устранить невозможно. Пациентам назначается симптоматическое лечение, психолого-педагогическая коррекция, реабилитационные мероприятия. Для уменьшения частоты эпилептических припадков используются антиконвульсанты, для нормализации сна – мелатонин. Занятия лечебной физической культурой и сеансы массаж направлены на развитие мелкой моторики и скоординированной походки, устранение сколиоза. Для улучшения коммуникативных навыков детей обучают языку жестов, вовлекают в групповые занятия, организуют сеансы поведенческой терапии, позволяющей освоить правила взаимодействия в обществе.
Продолжается поиск способов эффективного лечения синдрома. Проводится тестовое применение препаратов на генетически модифицированных мышах. Результаты доказывают, что ингибиторы топоизомеразы способны активировать материнский ген UBE3A. На данном этапе выполняются контрольные исследования, определяется безопасность и риски терапии, но информации пока недостаточно для перенесения экспериментов на группы людей.
На сегодняшний день еще не изобретен чудо-препарат, который бы помог победить это генетическое заболевание. Однако болезнь Ангельмана предполагает симптоматическую терапию, благодаря которой облегчается состояние пациента. При этом прописывается медикаментозное и немедикаментозное лечение. Синдром Петрушки у детей предусматривает такую терапию:
- Назначаются антиконвульсанты. Чаще прописывают Клоназепам, Конвулекс, Ламотриджин. Такие лекарственные средства сводят к минимуму частоту и интенсивность эпиприступов.
- Прописывают витаминотерапию (элементы групп B, C, D и E). Такое лечение укрепляет иммунную систему организма. Однако оно уменьшает эффективность противоэпилептических средств, поэтому все назначения должен делать доктор.
- Прописывается терапия, направленная на устранение проблем с пищеварительным трактом. Такое лечение предусматривает прием слабительных препаратов (Фитолакса или Сенаде) и пробиотиков (Хилак форте, Бифиформа).
- Назначают снотворные. Чаще пациентам прописывают Дифенгидрамин или Мелатонин.
- Показана гормональная терапия. Такое лечение направлено на коррекцию поведения пациентов. Например, гормон секретин улучшает процесс пищеварения, а окситоцин – увеличивают познавательные способности и память.
- В борьбе с проблемами суставов помогут физиопроцедуры. Назначаться могут парафиновые аппликации, массаж, магнитотерапия, электрофорез, аквагимнастика.
При этом заболевании показана поведенческая терапия, занятия с логопедом, психологом и дефектологом. У детей, у которых диагностирован синдром счастливой куклы, отмечаются серьезные нарушения речи. Некоторые из них имеют ограниченный словарный запас, а другим даже сложно воспроизводить отдельные звуки.
Занятия с такими детками должны быть непродолжительными (не более 30 минут), но регулярными: желательно, чтобы они проводились каждый день. По мнению специалистов, начинать можно с рисования пальчиковыми красками. Во время таких занятий ребенок успокаивается, знакомится с цветовой гаммой и развивает мелкую моторику.
Поскольку дети с синдромом Ангельмана лучше воспринимают информацию, когда она визуализирована, желательно при их обучении использовать карточки и другие наглядные пособия. Например, чтобы объяснить такому ребенку понятия «большой»» и «маленький», можно воспользоваться разборной матрешкой. При первом знакомстве малыша с таким наглядным пособием взрослый должен сам открыть игрушку и каждое свое действие прокомментировать. Затем можно попросить ребенка проделать то же самое. Постепенно задача усложняется: например, малыш должен показывать, где большая матрешка.
БРАТЬЯ И СЕСТРЫ

Мальчики и девочки становятся носителями синдрома Вильямса одинаково часто. Клиническая картина очень разнообразна. Симптомы можно разделить на необычный внешний вид, задержку в умственном развитии и различные физические особенности. Но больше всего Веронику удивил тот факт, что все заболевшие становятся похожими, как братья и сестры.
– Со временем у детей становится практически одинаковое строение лица. Это плоская переносица с округлым носом, увеличенный рот с приподнятыми вверх уголками, пухлые губы, полные щеки, небольшой заостренный подбородок, низко посаженные уши, выступающий затылок. Поэтому еще одно название недуга – синдром «лица эльфа», – поясняет Григорьева.
Все дело в том, что у детей с синдромом похожий паттерн лицевого строения, который заложен генетически. Кроме того, «вильямсята» значительно отстают от сверстников как в физическом, так и в интеллектуальном развитии. Сложности возникают с концентрацией внимания, организацией и планированием деятельности. У некоторых гиперактивность сочетается с импульсивностью и излишней коммуникабельностью.
При этом в младенческом возрасте дети отстают не критично. Они просто чуть позже начинают ходить или говорить, но навыки самообслуживания присутствуют: сами едят, одеваются, ходят в туалет.
– Некоторые даже могут освоить начальную школу, а вот в средней когнитивные способности уже, как правило, не позволяют учиться, – поясняет Григорьева.
Как ни странно, есть у синдрома и положительные стороны. Например, хорошая память на лица, большой словарный запас, способность к музыке.
– Такие дети преуспевают в задачах, связанных с разговорной речью, музыкой и механическим запоминанием. Есть люди с абсолютным слухом и чувством ритма. Плюс «вильямсята» чрезвычайно общительны, однако могут не понимать нюансы социального общения, – добавляет Григорьева. – Например, моя Маруся очень социально включенная. Разница между ней и сверстниками чувствуется, только если посадить их за одну парту и попросить читать или писать. Нельзя сказать, что она такая же, как все, но отличия минимальны.
КУДА ОБРАЩАТЬСЯ В ПОДМОСКОВЬЕ ПРИ ПОДОЗРЕНИИ НА ГЕНЕТИЧЕСКИЕ ОТКЛОНЕНИЯ?
Пройти обследование и попасть на прием к специалисту жители Подмосковья могут в Центре орфанных заболеваний. Учреждение в регионе появилось два года назад. Оно вошло в состав Московского областного консультативно-диагностического центра для детей в Мытищах. С тех пор маленькие пациенты с редкими наследственными заболеваниями получают здесь максимально точную диагностику, полноценное лечение и реабилитацию.Болезни Ниманна – Пика, Вильсона – Коновалова и Гоше, мукополисахаридоз, юношеский артрит с системным началом, несовершенный остеогенез, наследственный ангиоотек, гликогеноз – лишь малая часть редчайших генетических недугов, с которыми пациенты уже обращались в Центр. Учреждение занимается не только лечением пациентов, но и обучением новых специалистов и научно-практической работой.Адрес: Мытищи, ул. Коминтерна, д. 24 а, стр. 1.Телефон: 7 (498) 698-60-72.
К слову
Одной из самых редких болезней, которая встречается среди детского населения Московской области, является болезнь Гоше. В данный момент в регионе зафиксирован всего один несовершеннолетний с таким недугом. Болезнь Гоше – наследственное заболевание, при котором поражаются кроветворные органы, костная система. Оно серьезно сказывается на физическом развитии.
Лечение синдрома Вильямса — возможно ли?
Так как заболевание генетическое, специфического лечения на сегодняшний день не разработано. При выявлении пороков сердца или иных врождённых аномалий требуется хирургическая коррекция. Лечение в основном симптоматическое, то есть при каждом симптоме назначают определённый вид терапии. Артериальная гипертензия должна корректироваться лекарственными препаратами, например, из группы ингибиторов АПФ. Лечение подбирается индивидуально после проведения тщательного обследования и с учётом переносимости того или иного лекарственного средства. Пожизненно должна проводится коррекция гиперкальциемии (повышение концентрации кальция в крови) путём диетотерапии, из рациона следует исключать продукты, богатые витамином Д и кальцием, а также избегать приёма витамино-минеральных комплексов, в которые включены данные вещества
Работа с психологом и логопедом должна проводиться регулярно, также очень важно создать благоприятный психоэмоциональный фон в семье
Методы лечения
Медицина не располагает средствами, которые позволили бы вылечить полностью синдром Вильямса, поэтому в основе лечения лежит коррекция физических и умственных нарушений.
Чтобы уменьшить выраженность физических пороков (недоразвитость внутренних органов), назначается симптоматическая медикаментозная терапия, которая поддерживает работу сердца, почек, улучшает работу желудка и кишечника.
Препараты подбираются индивидуально в зависимости от особенностей и степени выраженности патологий. Часто необходимо проведение ряда оперативных вмешательств.
Специфическое медикаментозное лечение, направленное на коррекцию умственной отсталости и нарушений психики, не было разработано. Специалистами часто назначаются препараты из следующих групп:
- Ноотропные и нейрометаболические медикаменты (Церебролизин, Актовегин, Семакс). Защищают клетки мозга от гипоксии, улучшают адаптацию и обучаемость, положительно влияют на мозговое кровообращение.
- Седативные средства (Валериана, отвары успокоительных трав, Ново-пассит). Применяются, если ребенок проявляет повышенную тревожность. При развитии фобий и других психических отклонений показана работа с психотерапевтом, показано использование более сильных средств.
Также применяются следующие методы коррекции:
- Лечебная физкультура. Поддерживает мышечный корсет в тонусе, предотвращая возникновение искривлений позвоночного столба, улучшает моторику и координацию.
- Массаж. Положительно влияет на крепость мышц, улучшает координацию.
- Нейропсихологическая коррекция. В процессе специальных занятий ребенок учится обслуживать себя, разговаривать, читать и писать, расширяет словарный запас. Наилучший результат получается, если родители активно присоединяются к работе с ребенком и окружают его любовью.
Также детям показано соблюдение диеты, которая специально разрабатывается в зависимости от выраженности его отклонений.
В первые годы диетотерапия направлена на снижение концентрации кальция. Рекомендуется уменьшить количество соли и специй в блюдах, чтобы снизить риск развития артериальной гипертензии.
Что делать?
Если Вы считаете, что у вас Синдром Ангельмана
и характерные для этого заболевания симптомы, то вам могут помочь врачи: генетик, педиатр.
Желаем всем здоровья!
Заболевания со схожими симптомами
Синдром Мартина-Белл (совпадающих симптомов: 5 из 20)
Синдром Мартина-Белл – распространенная генетическая аномалия, которая чаще диагностируется у мальчиков, нежели у девочек. Данные медицинской статистики достаточно неутешительны – частота рождения малышей с таким недугом составляет 1 на 6000 девочек, и 1 на 4000 мальчиков.
Стоит отметить, что у представителей сильного пола заболевание протекает более тяжело, но все же бывают и некоторые исключения. Причиной проявления у девочек более стертой симптоматики является компенсирующий эффект, которые оказывает вторая Х-хромосома в кариотипе.
Примечания
- X-сцепленная умственная отсталость. (рус.). Центр Молекулярной генетики при Медико-генетическом научном центре РАМН. Проверено 8 мая 2020.
- Yamasaki K., Joh K., Ohta T., Masuzaki H., Ishimaru T., Mukai T., Niikawa N., Ogawa M., Wagstaff J., Kishino T. Neurons but not glial cells show reciprocal imprinting of sense and antisense transcripts of Ube3a. (англ.) // Human molecular genetics. — 2003. — Vol. 12, no. 8. — P. 837—847. — DOI:10.1093/hmg/ddg106. — PMID 12668607.
- Petersen M. B., Brøndum-Nielsen K., Hansen L. K., Wulff K. Clinical, cytogenetic, and molecular diagnosis of Angelman syndrome: estimated prevalence rate in a Danish county. (англ.) // American journal of medical genetics. — 1995. — Vol. 60, no. 3. — P. 261—262. — DOI:10.1002/ajmg.1320600317. — PMID 7573182.
- Steffenburg S., Gillberg C. L., Steffenburg U., Kyllerman M. Autism in Angelman syndrome: a population-based study. (англ.) // Pediatric neurology. — 1996. — Vol. 14, no. 2. — P. 131—136. — DOI:10.1016/0887-8994(96)00011-2. — PMID 8703225.
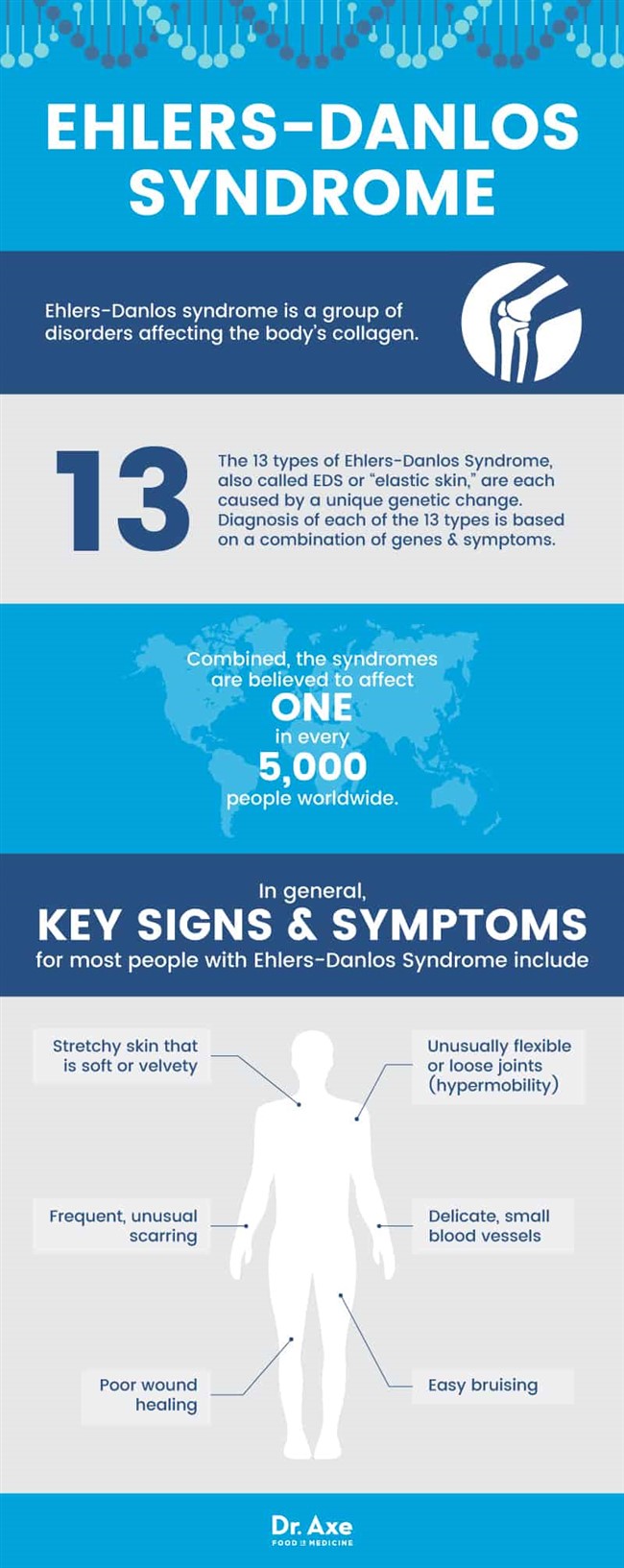
Формы синдрома Элерса-Данлоса
Болезнь имеет множество форм:
- Синдром Элерса-Данлоса Тип I (ТИП I ЭДС )
- Синдром Элерса-Данлоса Тип II (ТИП II ЭДС)
- Синдром Элерса-Данлоса III типа (тип ЭДС III и гипермобильности в суставах)
- другие формы (тип IV – тип X)
1 тип синдрома Элерса-Данлоса встречается чаще остальных (40-50%). При таком виде заболевания преобладают симптомы, затрагивающие кожный покров, возможность растяжения которого отходит от нормы в 2-2.5 раз. Данный тип сопровождается наружными кровотечениями и варикозными венами в области нижних конечностей, расшатанностью суставов тела, деформацией скелета. Роды при таком виде заболевания часто происходят преждевременно.
При 2 типе наблюдаются аналогичные признаки, но проявляются они не так сильно. Аномальная гибкость наблюдается только в суставах конечностей, преимущественно стоп и кистей. Растяжимость кожи незначительно отклоняется от нормы, то же и с нарушением работы кровеносных сосудов.
3 тип проявляется ввиду аутосомно-доминантного наследования и протекает доброкачественно. Включает повышенную подвижность суставов по всему телу, деформацию скелета и мышечных тканей и незначительные проявления эластичности кожи.
Редкий и тяжело протекающий 4 тип может быть доминантной и рецессивной. Отличается наличием, у больных повышенной подвижности суставов в пальцах рук, спонтанного возникновения гематом внутренних органов и разрывом всех видов сосудов, что часто приводит к летальным исходам.
5 тип синдрома Элерса-Данлоса происходит из-за X-сцепленного рецессивного наследования. Для него характерны умеренно повышенная мобильность суставов, кровоподтеки и повышенная растяжимость кожи и ее чувствительность.
6 тип наследуется по аутосомно-рецессивному типу. Помимо кровотечений, повышенной подвижности суставов и неестественно эластичной кожи характеризуется увеличением мышечного тонуса (гипертонией мышц), а так же косолапостью и тяжелой формой кифосколиоза, выявляется большой перечень патологий зрения.
7 тип, называемый артроклазией, может наследоваться аутосомно-доминантно и аутосомно-рецессивно. При этом типе у людей часто случаются травмы из-за нездоровой подвижности суставов. Пациентам, подверженным такому типу заболевания, свойственно иметь маленький рост.
8 тип характеризуется аутосомно-доминантным наследованием, и в большей степени для него характерна чувствительность кожи к внешним воздействиям, а так же воспаление тканей, окружающих зубы, что приводит к их ранней потере.
По итогам современных исследований 9 и 10 тип синдрома Элерса-Данлоса не входит в перечень типов классификации заболевания. Для них характерно аутосомно-рецессивное наследование. Кроме гиперэластичности кожи наблюдаются линейные полосы в местах, где кожа наиболее растянута.
Так же присутствует агрегация тромбоцитов и гиперподвижность суставов. У подверженных десятому типу часто наблюдаются врожденные вывихи бедра и постоянно повторяющиеся вывихи плечевых суставов и надколенника.
Симптомы синдрома Вильямса
Проявления синдрома Вильямса иногда могут не определяться при рождении ребенка – в этот период заподозрить наличие заболевания можно лишь по сниженной массе тела, врожденному подвывиху бедра (наблюдается не у всех больных), порокам сердца. Все эти факторы достаточно неспецифичны, поэтому чаще всего патологию обнаруживают в старшем возрасте. Одним из самых заметных симптомов синдрома Вильямса является характерный внешний вид лица больных («лицо эльфа») – плоская переносица с округлым носом, увеличенный рот с приподнятыми вверх уголками, пухлые губы, полные щеки, небольшой заостренный подбородок. Из других изменений в области лица и головы часто отмечают низко посаженные уши, выступающий затылок, эпикантус. Для больных синдромом Вильямса характерен голубой цвет глаз с выраженным рисунком на радужной оболочке, голубоватый оттенок склер, отечность верхних и нижних век, нередко – косоглазие.
 Геморрой в 79% случаев убивает пациента
Геморрой в 79% случаев убивает пациента
Из патологических изменений при синдроме Вильямса чаще всего регистрируются различные пороки сердца, что в некоторых случаях может привести к раннему летальному исходу. В основном эти нарушения сводятся к недостаточности различных клапанов. Также у больных синдромом Вильямса чаще, чем у здоровых детей, возникают пупочные и паховые грыжи. Для этого заболевания характерна генерализованная мышечная гипотония, обуславливающая вторичные нарушения формирования скелета – Х-образные ноги, сколиоз и другие искривления позвоночного столба, плоскостопие, деформации грудной клетки. При осмотре стоматолога у больных синдромом Вильямса выявляются длинные, но редко расположенные зубы, которые довольно часто поражаются кариесом и легко разрушаются.
Дети, страдающие синдромом Вильямса, значительно отстают от сверстников, как в интеллектуальном, так и в физическом развитии. Они имеют пониженную массу тела при рождении, медленно набирают ее в дальнейшем, их рост останавливается намного раньше, поэтому проявлением этого заболевания также является низкорослость. Масса тела после подросткового периода может увеличиваться, имеется риск развития ожирения. В интеллектуальном плане больные синдромом Вильямса на всю жизнь сохраняют признаки имбецильности (умеренная умственная отсталость, средний уровень IQ 40-80). При этом они достаточно легко идут на контакт, у них, как правило, хорошо развита устная речь, при правильной коррекционной работе они способны выполнять простейшие бытовые поручения. В некоторых случаях при синдроме Вильямса может наблюдаться эмоциональная неустойчивость, тревожность, энурез.
Отличительные признаки аномалии
- высокий, широкий лоб, непропорциональный зауженному книзу маленькому подбородку;
- плоская переносица переходит в короткий нос, у которого отсутствует кончик;
- разрез широко посаженных глаз узкий, цвет зрачка голубой, веки припухшие, сходящееся косоглазие;
- низко расположенные уши вытянутой кверху формы, что делает индивидуума похожим на персонажа фэнтези;
- рот большой, верхняя губа значительно тоньше нижней.
Кукольную внешность дополняют пухлые опущенные щеки, прямые, правильной формы брови.
У детей с этим диагнозом позже установленного нормой срока прорезаются зубы, деформируется челюсть, проявляются признаки неправильного прикуса. Отсутствие аппетита сказывается на росте, отмечается дефицит веса. Мышцы недостаточно развитые. К вторичной симптоматике синдрома относится плоскостопие, слабый вестибулярный аппарат, нарушение координации.
У человека в переходном возрасте признаки дополняются:
- заниженной линией талии;
- узкой грудной клеткой;
- непропорциональной туловищу длинной шеей;
- соединенными коленями.
Дефекты со стороны внутренних органов характеризуются:
- Выпячиванием внутрь стенки двенадцатиперстной кишки, что становится причиной частых запоров. Заболевание сопровождается хроническим гастритом.
- Формированием стеноза артерий и аорты, сердечной недостаточностью. Избыток кальция в организме усугубляет патологию.
- Недоразвитость щитовидной железы у плода становится причиной гормонального сбоя, появляются признаки сахарного диабета, гипотиреоза.
- Аномалией почек и мочевыделительной системы.
Малыши плохо читают, практически не владеют счетом. Невозможность обучения компенсируется хорошим словарным запасом, высокими коммуникативными способностями. В возрасте до четырех лет пациенты испытывают трудности в составлении осмысленного предложения, позже эта проблема самоустраняется.
Малыши в частых случаях обладают абсолютным слухом и чувством ритма. Легко знакомятся с людьми, у них хорошая память на лица. Привлекают внимания обширным словарным запасом и умением им пользоваться. Легко ведут беседу, доброжелательны, всегда позитивно настроены, улыбчивы и вежливы. Синдром Уильямса делает ребенка знаменитостью в кругу социума.
Взрослые отличаются инфантильностью, которая проявляется в несвойственных возрасту поступках, гиперактивностью, эмоциональной лабильностью. Необходимость в постоянном общении вызывает неадекватность поведения, формирует фобии. Для адаптации в социуме пациентам необходима помощь психолога и близких людей.
Факторы-провокаторы синдрома Вильямса
В научных кругах ведутся споры вокруг причины, вызывающей генетическое заболевание. Часть специалистов поддерживает теорию наследственного фактора, остальные склоняются к спонтанной мутации во время зачатия. Механизмом, запускающим патологический процесс, является делеция (утрата) пары в седьмой хромосоме. В состав отсутствующей копии входит больше 25 кодирующих генов, от которых зависит:
- углеводный метаболизм;
- синтез эластина;
- функционирование головного мозга;
- кальциевый обмен;
- состояние нейронов в нервной ткани;
- развитие интеллекта.
Недостаточное производство эластина, входящего в клеточный состав стенок сосудов и сердечного клапана, приводит к появлению пороков органа и гипертензии. Нарушение метаболизма кальция становится причиной гиперкальциемии, при которой истончаются стенки артерий и клапана, вызывая сердечно-сосудистую недостаточность. Дефицит питания нейронов отражается на формировании центральной нервной системы, развитии головного мозга, лицевой дисморфологии.
Вероятность синдрома Вильямса у потомства возрастает, если генетическая мутация диагностировалась у родителей или близких родственников. В этом случае не отмечено выпадение одной копии в наборе на момент зачатия. С учетом клинических проявлений у новорожденного недостающее звено в седьмой хромосоме плод получил путем наследования. Такая причина развития аномалии встречается очень редко.
Более распространенная теория, чем аутосомно-доминантный вариант, утверждает, что синдром Вильямса у детей формируется путем спонтанного нарушения хромосомной цепочки в момент зачатия. Провоцирующим фактором является воздействие на организм одного или обоих родителей:
- вида трудовой деятельности на вредном производстве, связанном с ядовитыми химикатами, тяжелыми металлами, радиацией;
- концентрация токсинов в организме по вине плохой экологии в месте проживания мужчины или женщины;
- употребление алкоголя, наркотиков, курение табака.
В этом случае у родителей существует риск появления на свет ребенка с недостающим звеном в хромосоме.
ДИАГНОЗ
Синдром Вильямса – редчайшая генетическая патология – один случай на 15–20 тыс. новорожденных. При заболевании в организме ребенка теряется от 25 до 29 генов. Это приводит, например, к порокам сердца, хриплому голосу, ортопедическим проблемам, изменениям черт лица.
Диагноз может подтвердить лишь врач-генетик, к которому обычно направляют из поликлиники, если замечают странности.
– В нашем случае что-то неладное заподозрил кардиолог, – рассказывает Вероника. – Когда Маше был год, у нее диагностировали порок сердца
При этом специалисты обратили внимание на внешность и направили к генетику. Так я впервые узнала о существовании болезни
Впервые синдром Вильямса был описан в 1961 году кардиологом из Новой Зеландии Джоном Вильямсом. Он заметил, что у некоторых детей с сердечными проблемами имеются схожие внешние признаки. Вильямс также доказал генетическую природу этого недуга.